Pachyonychia Congenita type 1 – A peerless entity
Yugandar Inakanti1, Venkata Narsimha Rao Thimmasarthi2, Shiva Kumar1, Akshaya Nagaraj1, Srilakshmi Peddireddy1
1Department of Dermatology, Venerology and Leprosy, P.E.S. Medical College, Kuppam, India, 2Department of Dermatology, Venerology and Leprosy, Guntur Medical College, Guntur, India
ABSTRACT
Pachyonychia congenita (PC) is a rare Autosomal dominant genodermatosis characterized by hyperkeratosis affecting the nails and palmoplantar areas, oral leucokeratosis, and cystic lesions. Although classically subdivided into two major variants, PC-1 (Jadassohn-Lewandowski syndrome) and PC-2 (Jackson-Lawler syndrome), according to the localization of the mutations in the KRT6A/KRT16 or KRT6B/KRT17 genes, respectively. We report a 10 year-old male patient with a history of thickened, discoloured nails, raised spiny skin lesions all over the body since birth with focal plantar keratoderma and absence of natal teeth.
Key words: Jadassohn-lewandowsky syndrome; keratins; nails; pachyonychia congenita; palmoplantar keratoderma
INTRODUCTION
Pachyonychia congenita (PC) describes a group of rare autosomal dominant skin disorders characterized predominantly by dystrophic, thickened nails and painful and highly debilitating palmoplantar hyperkeratosis [1,2]. With a base on phenotypes, PC is classified in four types: PC I to III and PC tarda [3,4]. The objective of this case report is to describe the clinical features of PC-I.
CASE REPORT
A 10 year-old male born at preterm to non consanguineous parents was referred to our outpatient clinic for evaluation of thickened nails starting at the age of 6 months.
The boy had been born prematurely at 34 weeks’ gestation after premature contractions and prolonged rupture of membranes. No maternal fever, tachycardia, chorioamnionitis, or other complications had occurred. The boy had stayed in neonatal intensive care for 4 weeks because of feeding issues, and he had been transiently on nasal continuous positive airway pressure therapy, from which he was weaned to room air after 3 days of life. He was discharged at 38 weeks’ gestation. He presented with history of pinhead-sized yellowish spiny skin lesions all over body noted at birth that had slowly grown during the first year of age and abnormalities nails. No history of natal or neonatal teeth. The patient intelligence was normal. No similar complaints in family.
Physical examination showed:
1. All 20 nails were thickened, yellowish discolouration with increased curvature and subungual hyperkeratosis (Fig 1).
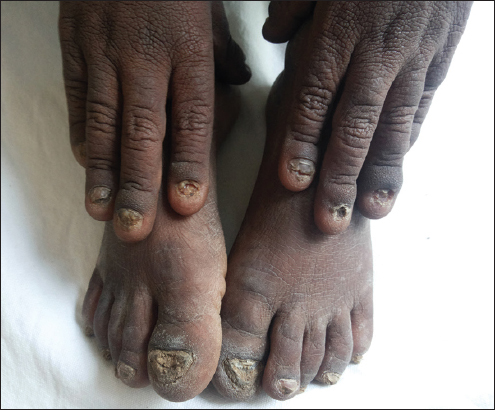
Figure 1: 20 thickened nails, yellowish discolouration with increased curvature and subungual hyperkeratosis.
2. All toe nails were thickened, dystrophy, yellowish discolouration and subungual hyperkeratosis (Fig. 2).
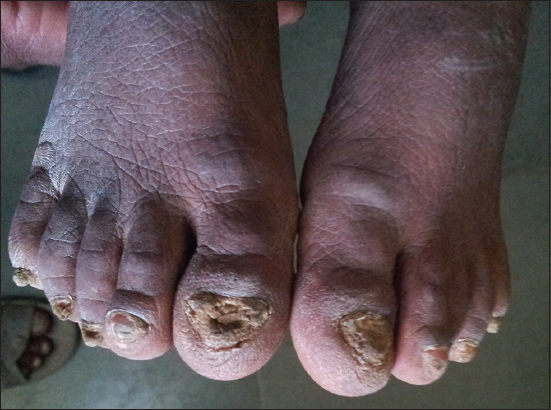
Figure 2: 20 thickened nails, yellowish discolouration with increased curvature and subungual hyperkeratosis.
3. Skin: Multiple follicular discrete papules present over the face, neck and upper chest (Fig. 3).
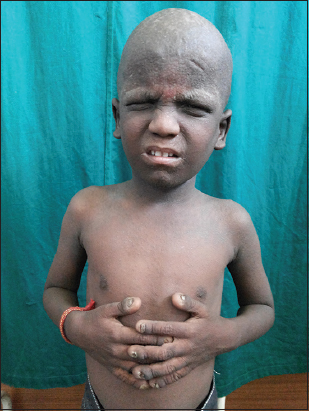
Figure 3: Multiple follicular discrete papules present over the face, upper chest, buttock, elbow, knees and flexures.
4. Multiple hyperkeratotic papules present over the back of neck, back of chest, buttock and elbow (Fig. 4).
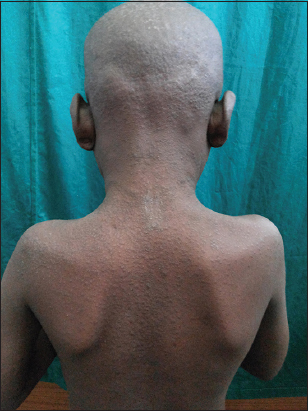
Figure 4: Multiple follicular discrete papules present over the face, upper chest, buttock, elbow, knees and flexures.
5. Focal plantar keratoderma (Fig. 5).
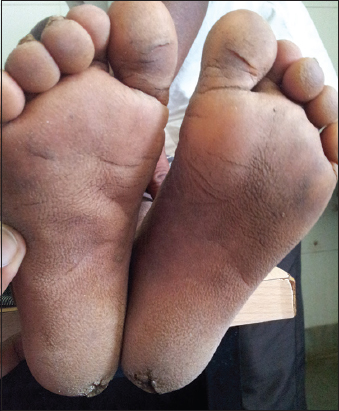
Figure 5: Focal plantar keratoderma.
There were no abnormalities in examination of otolarynx and ophthalmology.
All laboratory investigations were within normal limits. KOH mounts of toe and finger nails were negative for fungal elements. Histopathology examination of Follicular keratotic papule showed follicular plugging, hyperkeratosis and acanthosis. Skin biopsy of Sole showed marked hyperkeratosis, acanthosis, moderate hyper granulosa’s and minimal dermal inflammatory infiltration. Based on characteristic clinical findings and histopathology features, diagnosis of pachyonychia congenital type1 or Jadassohn – Lewandowsky syndrome.
Pachyonychia congenita (PC) is a rare, autosomal dominant disorder characterized by triad of subungual hyperkeratosis with accumulation of hard keratinous material beneath the distal portion of the nails, lifting the nails from the nail bed, keratosis palmaris et plantaris with thick callosities, especially on the soles and thick white areas on the oral mucosa [5]. Other associated features which may occur include keratosis pilaris, hyperkeratotic follicular papules on the sites of friction, hair abnormalities and hyperhidrosis of the palms and soles. These disorders have been suggested to be due to mutations in paired keratins, K6a/K16 (in PC1) and K6b/K17 (in PC2). According to these mutations, various clinical variants have been described:
PC-I or Jadassohn – Lewandowsky syndrome (MIM #167200) is a common entity characterized by onychogryposis on all the digits (100%), hyperkeratosis of the palmo plantar (50-90%) and of extensor areas, follicular keratosis (37%), oral (50-75%) or laryngeal (6-15%) leukokeratosis, acral hyperhidrosis (20-75%) and blisters (36%) [2–7,8,9].
In patients with PC-II or Jackson- Lawler syndrome (MIM #167210) main changes are natal or neonatal teeth (15-50%), cutaneous cystic lesions (25%), disorders in scalp and eyebrow hairs (9-25%), in addition to corneal dystrophy (8%); moreover, the nail hyperkeratosis is less accentuated in PC-II than in PC-I [3,6,7,8,9].
PC-III (Schafer-Brunauer syndrome) shows ccombined features of type 1 and 2 with angular cheilosis, cataract and corneal dyskeratosis. [4]. Despite recent advances in genetic analysis, Misdiagnosis can occur because same mutations may show diverse features [10].
A fourth variant, PC tarda, has also been described and is characterized by a later onset that ranges from late childhood to middle ages.
Other rare variants include pachyonychia congenital with only nail involvement. Patients with PC often present at birth or soon after with the characteristic hypertrophic toenail dystrophy [11].
Pathogenesis
Keratins are structural proteins that promote the integrity of epithelial cells. As a result mutations in the genes encoding keratins lead to cell fragility [1,12]. The skin expresses the largest number of keratin genes of any organ in body. PC is caused by mutations in 5 keratin genes KRT6a, KRT6b,KRT6c, KRT16 and KRT17 which are expressed only in palmo plantar skin, the nail bed, pilosebaceous unit and oral mucosa, leading to selective involvement of these sites in PC [1,13,14].
Genetic Diagnosis
PC is an autosomal dominant disorder, which has been reported worldwide with approximately equal prevalence in males and females. More than 45% of cases appear spontaneously with no family history of PC [11]. Given the overlapping clinical presentation with other genetic disorders, only genetic testing can confirm the PC diagnosis.
With nearly 100 distinct PC mutations now identified, correlating the signs of PC with specific mutations and genes has led to a new classification system of PC. While in the past, PC has been classified according to phenotypic features into PC-l and PC-2, the disorder is now classified into 5 subgroups corresponding to the underlying genetic defect: PC-K6a, PC-K6b, PC-K6c, PC-K16 and PC-K17 [11,13].
Histological Findings
Histological examination of plantar hyperkeratosis plaques reveals an acanthosis epidermis with parakeratosis and orthokeratosis compatible with rapid keratinocyte proliferation and differentiation. Cytologic atypical is not seen.
Immunostaining shows positive immune staining of K14, as would be expected along with K6, K16, K17 in the basal cell layer. In the supra basal layers, K6, K16, K17, and K14 staining persists and K10 staining appears [14,15].
Electron microscopy on palmar or plantar plaques shows thickened and clumped intermediate filaments, as well as enlarged keratohyaline granules. In the broadened granular layer, thick masses of monofilaments and large, irregular keratohyaline granules are present. In the spinous layer, thick masses of monofilaments are found at the periphery of the cells.
Complications like respiratory distress due to laryngeal leucokeratosis and acroosteolysis, malignant changes in palmoplantar lesions can occur in pachyonychia congenital.
Treatment
Treatment options for PC fall into four broad categories:
- Non- invasive (mechanical) e.g. abrasion with some hand tool
- Invasive (surgical) e.g. electrofulgration, excision
- Chemical methods using urea, propylene glycol, alpha hydroxy acid
- Pharmacological (vitamin A, retinoid), all basically targeted at reducing the hyperkeratosis involving different sites [16].
When the familial mutation is known, genetic counselling can be done and if required, prenatal diagnosis can be done at early stage of pregnancy by chorionic villi biopsy. In our case we advised keratolytics and oral isotretinoin.
CONCLUSION
PC is a rare genetic disorder for which there are very few therapeutic options are available. Free genetic testing is provided to each patient through the International Pachyonychia Congenita Research Registry(IPCRR) sponsored by the PC Project (www.pachyonychia.org) which is a non-profit USA public charity, supports clinical and research activities related to the treatment of pachyonychia congenital. By building a patient community through the IPCRR and a physician and researcher community through the IPCC, PC Project is moving research forward to better understand the condition and to develop effective treatments.
In India we don’t have such projects working on PC. With this case, we intend to draw attention to this condition and the role of the dermatologist in the diagnosis. To our best of the knowledge this represents the first report of isolated PC in siblings with healthy parents.
ACKNOWLEDGEMENT
We gratefully acknowledge the help of the
- Principal, PESIMSR, Kuppam.
- The professor and head of the department of DVL, PESIMSR, Kuppam.
CONSENT
The examination of the patient was conducted according to the Declaration of Helsinki principles. Written informed consent was obtained from the patient for publication of this article.
REFERENCES
1. Leachman SA, Kaspar RL, Fleckman P, Florell SR, Smith FJ, McLean WH, Clinical and pathological features of pachyonychia congenitaJ Investig Dermatol Symp Proc 2005; 10: 3-17.
2. McLean WH, Hansen CD, Eliason MJ, Smith FJ, The phenotypic and molecular genetic features of pachyonychia congenitaJ Invest Dermatol 2011; 131: 1015-17.
3. Al Aboud A, Al Aboud K, Josef Jadassohn (1863-1936), Felix Lewandowsky (1879-1921), and their syndromeClin Cosmet Investig Dermatol 2011; 4: 179-82.
4. Ehsani A, Moeineddin F, Rajaee A, Pachyonychia congenita with woolly hair in a ten month old infantIndian J Dermatol Venereol Leprol 2008; 74: 485-6.
5. Johnson BL, JrYan AC, Elder DE, Congenital diseases (Genodermatosis)Lever’s Histopathology of the Skin 2009; 10th edition. Philadelphia: Lippincott Williams & Wilkins; 138
6. Balasubramanian S, Kaarthigeyan K, Ramnath B, Pachyonychia congenita with unusual featuresIndian Pediatr 2009; 46: 897-9.
7. Karen JK, Schaffer JV, Pachyonychia congenita associated with median rhomboid glossitisDermatol Online J 2007; 13: 21
8. Roche-Gamón E, Mahiques-Santos L, Vilata-Corell JJ, Paquioniquia congénitaPiel 2006; 21: 72-8.
9. Rodríguez H, Cuestas G, Zanetta A, Pachyonychia congenita with involvement of the larynxActa Otorrinolaringol Esp 2013; 6519: 264-6.
10. Vaccaro M, Guarneri F, Barbuzza O, Guarneri C, Pachyonychia congenita tarda affecting only the nailsDermatol Online J 2008; 14: 12
11. Eliason MJ, Leachman SA, Feng BJ, Schwartz ME, Hansen CD, A review of the clinical phenotype of 254 patients with genetically confirmed pachyonychia congenitaJ Am Acad Dermatol 2012; 67: 680-6.
12. Coulombe PA, Lee CH, Defining keratin protein function in skin epithelia: epidermolysis bullosa simplex and its aftermathJ Invest Dermatol 2012; 132: 763-75.
13. Wilson NJ, Leachman SA, Hansen CD, McMullan AC, Schwartz ME, McLean WH, A large mutational study in pachyonychia congenitaJ Invest Dermatol 2011; 131: 1018-24.
14. Wilson NJ, Messenger AG, Leachman SA, O’Toole EA, Lane EB, McLean WH, Keratin K6c mutations cause focal palmoplantar keratodermaJ Invest Dermatol 2010; 130: 425-9.
15. Wollina U, Schaarschmidt H, Fünfstück V, Knopf B, Pachyonychia congenita. Immunohistologic findingsZentralbl Pathol 1991; 137: 372-5.
16. Milstone LM, Fleckman P, Leachman SA, Leigh IM, Paller AS, van Steensel MA, Treatment of pachyonychia congenitaJ Investig Dermatol Symp Proc 2005; 10: 18-20.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Comments are closed.