Antifungal drugs and resistance: Current concepts
Pramod Kumar Nigam
Department of Dermatology, STD & Leprosy, Pt. J. N. M. Medical College, Raipur-492001, (CG), India
ABSTRACT
Recently, clinical failure and relapses have been observed in patients treated with antifungals. Drug resistance has become an important problem leading to significant negative social, psychological, and occupational health effects and quality of life. Early recognition and treatment is essential to reduce morbidity and possibility of transmission. The increased use, inappropriate prescribing and over the counter sale of antifungal agents has also added in the development of resistance to these drugs. The main biochemical and molecular mechanisms that contribute to antifungal resistance include reduced uptake of the drug, an active transport out of the cell or modified drug metabolic degradation of the cell, changes in the interaction of the drug to the target site or other enzymes involved in the process by point mutations, overexpression of the target molecule, overproduction or mutation of the target enzyme, amplification and gene conversion (recombination), and increased cellular efflux and occurrence of biofilm. Although, there is considerable knowledge concerning the biochemical, genetic and clinical aspects of resistance to antifungal agents, expansion of our understanding of the mechanisms by which antifungal resistance emerges and spreads, quicker methods for the determination of resistance, targetting efflux pumps, especially ATP binding cassette (ABC) transporters and heat shock protein 90, new drug delivery systems, optimizing therapy according to pharmacokinetic and pharmacodynamic characteristics, new classes of antifungal drugs that are active against azole-resistant isolates, and use of combinations of antifungal drugs or use of adjunctive immunostimulatory therapy and other modalities of treatment will clearly be important for future treatment strategies and in preventing development of resistance.
Key words: Antifungal, Resistance, Mechanism, Efflux pump, Measures
INTRODUCTION
Superficial dermatophytoses affecting skin, hair and nail are among the most common public health problem in hot and humid climate of tropical countries like India. Cutaneous mycoses are mostly caused by keratinophilic filamentous fungi called dermatophytes and are classified into three genera: Trichophyton, Microsporum and Epidermophyton. So far, about 30 species of dermatophytes have been identified as human pathogens [1]. Although infections caused by dermatophytes are generally limited to the surface regions of the skin, these fungi can behave in a manner invasive, causing deeper and disseminated infection, especially in immunocompromised patients [2]. World Health Organization estimates dermatophytes affect about 25% of the world population [3]. It is also estimated that 30 to 70% of adults are asymptomatic carriers of these pathogens, and that the incidence of this disease increases with age [3]. The estimated life-time risk of acquiring dermatophytosis is between 10 and 20 percent [4]. Climatic factors, as well as social practices, population migration and individual characteristics, such as immune status, may affect the epidemiology of dermatophytosis [3]. In addition, some risk factors have also been associated with onychomycosis, such as age, morphological abnormalities in the nails, genetic factors, poor hygiene conditions and some diseases such as diabetes mellitus and immunodeficiency frames [4,5].
The most common dermatophytes that cause cutaneous mycoses are Trichophyton rubrum, Trichophyton mentagrophytes, Microsporum canis and Trichophyton tonsurans [6]. Among the fungi isolated from skin infections, the anthropophilic dermatophyte T. rubrum is the most frequent amongst clinical cases of tinea pedis, tinea unguium, tinea corporis and tinea cruris [4]. while Trichophyton tonsurans was the most likely etiologic agent in cases with tinea capitis [7]. T. rubrum accounted for 76 percent of all superficial fungal diseases in a representative sample of the U.S. population [7]. Epidemiological studies on occurrence of dermatophytes have also shown that T. rubrum is present in 80% of cases and T. mentagrophytes in 20% [8]. Among onychomycosis also, T. rubrum is the most prevalent dermatophyte and affects children and adults in about 33.2% of the cases identified, followed by T. mentagrophytes in 6.3% cases [9]. In a study conducted over a period of three years, Bright et al reported the isolation of dermatophytes in 12.99% of cases of onychomycosis, and T. rubrum isolated in 9.04% of patients and T. tonsurans and T. mentagrophytes 2.54% and 1.41% of subjects, respectively [10].
Mycoses may have significant negative social, psychological, and occupational health effects and can compromise the quality of life significantly. Early recognition and treatment is essential to reduce morbidity and possibility of transmission. Treatment of dermatophytosis is generally long and costly. Dermatophytoses are often associated with relapses following the interruption of antifungal therapy. Recently, clinical failure has been observed in patients treated with antifungals and drug resistance has become an important problem. Although the prevalence of drug resistance in fungi is below that observed in bacteria, mycologists now believe that selective pressure will, over time, lead to more widespread resistance [11].
ANTIFUNGAL DRUGS AND THEIR MECHANISM OF ACTION
The fungal cell wall is composed of multiple layers where mannoproteins and glucan make up more than 80% of the cell wall composition, while chitin represents less than 2% [12]. Mannoproteins are predominantly expressed at the external surface. The plasma membranes of fungi are primarily composed of ergosterol [12]. Keratinocytes are the most numerous cells in the epidermis, forming a physical barrier to micro-organisms and mediate the immune response [3]. Keratinocytes secrete various soluble factors capable of regulating the immune response, such as growth factors [basic fibroblast growth factor (bFGF), transforming growth factor (TGF- α, TGF- β), tumor necrosis factor, interleukins (IL- 1, IL-3, IL-6, IL-7, IL8), and colony stimulating factors [13].
Recent studies have demonstrated that keratinocytes have a different profile of cytokine expression when stimulated by different species of dermatophytes [3]. It was shown that Arthroderma benhamiae, a zoophilic and teleomorph of dermatophyte T. mentagrophytes, induces the expression of several cytokines by keratinocytes which may be involved in triggering an inflammatory response typical of these infections [14]. Both topical and systemic therapies may be used to treat dermatophyte infections. The binding and synthesis of ergosterol, the major cell membrane component, are the targets for several antifungal structures. Topical therapy is generally effective for uncomplicated tinea corporis of small areas and of short duration [15].
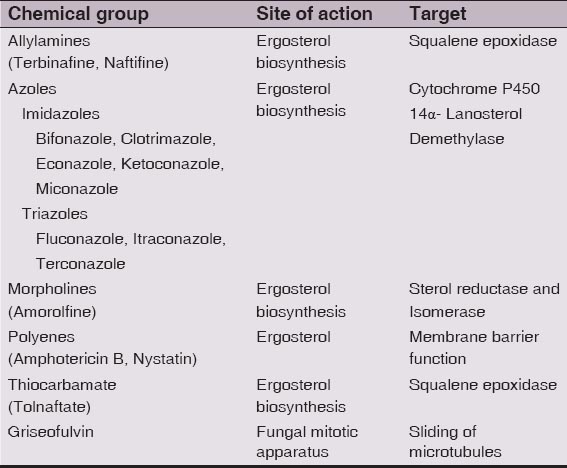
Antifungals are grouped on the basis of their structure and mechanism of action: The azoles and triazoles interfere with the ergosterol biosynthesis pathway by inhibiting cytochrome P450-dependent 14–demethylase and blocking the oxidative removal of 14–methyl from lanosterol resulting into the structural changes in the lipid membrane. They are directed against lanosterol 14-alpha-demethylase, a cytochrome P-450 enzyme containing a heme moiety in its active site, in the ergosterol pathway. Azoles have also been reported to inhibit membrane-surface enzymes and lipid biosynthesis [12]. Azoles, include imidazoles (ketoconazole and miconazole) and triazoles (fluconazole, itraconazole, and voriconazole). The topical azoles include clotrimazole, miconazole, oxiconazole, sulconazole, econazole, ketoconazole, sertaconazole. Sertaconazole is fungicidal and has anti-inflammatory and anti-itch properties [16]. Luliconazole is another FDA approved topical imidazole for treatment of interdigital tinea pedis, tinea cruris, and tinea corporis [17]. Recently, US Food and Drug Administration (FDA) approved First Topical Triazole, efinaconazole 10% topical solution, for the treatment of onychomycosis [18]. Polyenes include amphotericin B and nystatin. They increase the permeability of the plasma membrane. They bind to fungal membrane sterol, resulting in the formation of aqueous pores through which essential cytoplasmic materials leak out and thereby destroying the proton gradient within the membrane [19,20]. Allylamines (naftifine, terbinafine and the related benzylamine butenafine) and thiocarbamates (tolnaftate and tolciclate) inhibit the conversion of squalene to 2,3-oxidosqualene by the enzyme squalene epoxidase [21–23]. This enzyme blocks ergosterol biosynthesis, leading to intracellular accumulation of squalene which is toxic to fungal cells and leads to cell death [21]. Allylamines are lipophilic, bind effectively to the stratum corneum and also penetrate deeply into hair follicles [24]. Ciclopirox olamine is a topical fungicidal agent. It causes membrane instability by accumulating inside fungal cells and interfering with amino acid transport across the fungal cell membrane [25]. Candins function by inhibiting the synthesis of α 1,3-glucan which is the major structural polymer of the cell wall [26]. Echinocandins are semisynthetic lipopeptides that competitively inhibit α-glucan synthetase; the mechanism of action is not well defined but does not involve cytochrome P450 inhibition or P-glycoprotein transport [27]. Flucytosine inhibits macromolecular synthesis [28]. Morpholines (fenpropimorph and amorolfine) are recently introduced new class of antifungal drug for topical use. The morpholines inhibit two enzymes in the ergosterol biosynthetic pathway, C-14 sterol reductase (ERG24) and C-8 sterol isomerise (ERG2) [29]. As it acts on two different enzymes involved in sterol biosynthesis leading to inhibition of ergosterol biosynthesis in the fungal cell membrane thus making it a potent fungistatic and fungicidal agent. Alteration in the membrane sterol content leads to changes in membrane permeability and disruption of fungal metabolic processes [30,31]. In vitro studies have demonstrated that at concentrations of 0.1-100 μg/ml, topical amorolfine induces varying degrees of damage to the nuclear, mitochondrial and plasma membranes of both T mentagrophytes and Candida albicans [32]. An in vitro study has shown topical amorolfine to have the lowest minimum inhibitory concentration (MIC) against various strains of dermatophytes as compared to other topical antifungal agents [33]. Thus, azoles, allylamines, and thiocarbamates, and morpholines act through inhibition of the ergosterol biosynthetic pathway by interacting with enzymes involved in the synthesis of ergosterol from squalene.
Systemic therapy may be indicated for tinea corporis that includes extensive skin infection, immunosuppression, resistance to topical antifungal therapy, and comorbidities of tinea capitis or tinea unguium or when the infection involves hair follicles, such as Majocchi granuloma. The preferred treatment for tinea imbricata is griseofulvin or terbinafine, although some resistance has developed to oral griseofulvin [34]. The mode of action of griseofulvin is not completely clear, but it has been speculated that griseofulvin inhibits microtubule binding within the mitotic spindle in metaphase, causing arrest of fungal cell mitosis, weakening the cell structure [20]. A dose of 10 mg/kg/d is effective. In addition, griseofulvin induces the cytochrome P-450 enzyme system and can increase the metabolism of CYP-450–dependent drugs [25]. It is the systemic drug of choice for tinea corporis infections in children [25]. Oral terbinafine may be used at a dosage of 250 mg/d for 2 weeks; the potential exists for cytochrome P-450, specifically CYP-2D6, drug interactions with this agent [34]. Systemic azoles [eg, fluconazole (50-100 mg/d or 150 mg once weekly; itraconazole (100 mg/d); ketoconazole (3-4 mg/kg/d)] function similar to the topical agents, causing cell membrane destruction [34]. Based on E-test for susceptibility of T rubrum, voriconazole was the most active and fluconazole was the least active of the azole drugs [35]. Use of oral agents requires attention to potential drug interactions and monitoring for adverse effects [25]. Voriconazole and Posaconazole are two broad spectrum triazole antifungal agents that were recently approved. Ravuconazole [36] is a new member of the azole family and Pramiconazole is another new member of triazole class in the stages of development for the treatment of superficial infections caused by dermatophytes, yeasts and many other fungi [37].
FUNGAL RESISTANCE AND MECHANISMS OF ANTIFUNGAL RESISTANCE
Dermatophytoses are frequently associated with relapses following the interruption of antifungal therapy. Clinical resistance to antifungal agents was rare until the late 1990s, with only isolated cases in patients with chronic mucocutaneous candidiasis [38,39]. The incidence of fungal infections, including resistant infections, has increased during the last few years, and may be due to inadequate or irregular use of drugs or increased incidence of immunodeficiency states [38,40]. The increased use and over the counter sale of antifungal agents in recent years has also resulted in the development of resistance to these drugs. Drug resistance in fungi, especially to azoles, is becoming more prevalent clinically. After the appearance of resistance to griseofulvin, a case of clinical resistance to terbinafine was reported in 2003 [3]. Antifungal-drug resistance is usually quantified using the minimum inhibitory concentration (MIC), in which growth in the presence of a range of drug concentrations is measured over a defined time period according to a standard protocol [41]. The lowest drug concentration that results in a significant reduction of growth (usually either 50% or 90% reduction of growth compared with growth in the absence of the drug) is called the MIC. One well-known limitation of the MIC as a measure of resistance is that it does not always predict the clinical outcome of antifungal therapy [42]. In 1997, the NCCLS proposed methods and guidelines for antifungal resistance testing of yeasts [43]. The NCCLS method for the determination of antifungal susceptibility is currently a standardized technique that can be used to determine the levels of resistance of a yeast strain. The NCCLS document M38-P proposes methods and guidelines for resistance testing of filamentous fungi. There is currently no susceptibility standard for dermatophytes [44]. For aid in clinical interpretation of antifungal susceptibility testing, the NCCLS Subcommittee for Antifungal Susceptibility Testing recently established interpretive breakpoints for testing of fluconazole and itraconazole for Candida infections [45].
In vitro demonstration of resistance does not necessarily equate to in vivo resistance [44,46]. Other determinants in the selection of resistance include host-related factors, e.g. immunosuppression, the site and severity of infection and drug pharmacokinetics [47].
Martinez-Rossi described the antifungal resistance mechanisms in dermatophytes [48]. The main biochemical and molecular mechanisms that contribute to antifungal resistance include reduced uptake of the drug, an active transport out of the cell or modified drug metabolic degradation of the cell, changes in the interaction of the drug to the target site or other enzymes involved in the same enzymatic process by point mutations, overexpression of the target molecule, overproduction or mutation of the target enzyme, amplification and gene conversion (recombination), and increased cellular efflux [48,49].
Ergosterol is the predominant component of the fungal cell membrane [50] and serves as a bioregulator of membrane fluidity and consequently of membrane integrity in fungal cells [51]. Changes in the sterol and/or the phospholipid composition of the fungal cell membrane and membrane fluidity may result into a decrease in azole uptake by the fungal cell. Similarly, reduced intracellular accumulation of the drug may occur due to increased active transport of the drug out of the cell. Azoles, polyenes, allylamine and thiocarbamates owe their antifungal activities to inhibition of synthesis of or direct interaction with ergosterol. There are as yet no reports of modification of azole antimicrobials as a mechanism of resistance [28]. Resistant strains either exhibit a modification in the quality or quantity of target enzyme, reduced access to the target, or a combination of these mechanisms [28]. Various mechanisms by which microbial cells might develop resistance include [28]. 1. The target enzyme is overproduced, so that the drug does not inhibit the biochemical reaction completely. 2. The drug target is altered so that the drug cannot bind to the target. 3. The drug is pumped out by an efflux pump. 4. The entry of the drug is prevented at the cell membrane/cell wall level. 5. The cell has a bypass pathway that compensates for the loss-of-function inhibition due to the drug activity. 6, Some fungal enzymes that convert an inactive drug to its active form are inhibited. 7. The cell secretes some enzymes to the extracellular medium, which degrade the drug.
Several studies observed an alteration in the quantity or quality of 14α-demethylase in the expression of resistance to azole antifungal agents [28,52]. A recent study on resistance to fluconazole by comparing sterol composition, fluconazole accumulation, and inhibition of 14α-demethylase by fluconazole, no significant differences in the sterol content of C. krusei and C. albicans were detected [52,53]. Additionally, the enzyme had a low binding affinity for azole antifungals [54]. Overexpression of 14α-demethylase has also been implicated as a mechanism of resistance to azole antifungals [55]. However, Ghannoum and Rice suggest that overexpression of target enzyme plays only a limited role in clinical resistance to the azoles [28].
The resistance of dermatophytes to agents inhibitors involves the participation of modifiers target enzymes, overexpression of ABC transporters and stress-related proteins [48]. In T. rubrum two ABC transporters, like TruMDR1 and TruMDR2 were identified as playing important role in development of resistance to many antifungal process and also in the secretion of enzymes [56,57].
A role of upregulation of the ERG11 gene, which encodes the major target enzyme of the azoles lanosterol 14α–demethylase, has been observed in azole-resistant C. albicans and C. glabrata isolates [58,59]. However, other studies have reported no significant change in expression levels of the ERG11 gene in azole resistant clinical isolates of C. glabrata [60,61]. White [62] investigated the target enzyme (Erg11p) susceptibility to fluconazole in cell extracts and observed that a substantial decrease occurred in one of the isolate, corresponding to resistance development. Sequence analysis identified a single point mutation that resulted in a single-amino-acid substitution ‘R467K’ [63]. A second significant change observed in the ERG11gene of the resistant isolate was reported by White [63], namely, loss of allelic variation in the ERG11 promoter and in the downstream THR1 gene (which encodes homoserine kinase and is involved in threonine synthesis). The affinity of fluconazole for lanosterol 14α–demethylase containing the mutations Y132H, G464S or R467K was reduced as compared with the wild-type enzyme, confirming that these naturally occurring mutations indeed caused drug resistance in clinical C. albicans isolates [64,65]. Although these changes may account for resistance development, they are not the only factors involved and overexpression of the ERG11 gene probably is not critical for the development of azole resistance [66].
A mutation in the gene encoding the enzyme squalene epoxidase target antifungal terbinafine and gave high resistance to this drug against fungi T. rubrum [67]. Recently, Walsh et al [68] suggested that C. albicans may possess one or more additional genes encoding ATP-binding cassette MDR-like proteins that are distinct from CDR1, which could participate in the development of azole resistance. In this regard, five CDR genes (CDR1 to CDR5) which belong to the PDR family have been identified in C. albicans [69,70].
Considerable evidence implicating drug efflux as an important mechanism of resistance to azole antifungals is forthcoming recently. Studies [71–74] indicate that fungi possess at least two efflux systems: (i) proteins belonging to the major facilitator superfamily (MFS) and (ii) ATP-binding cassette (ABC) superfamily of proteins. The MFS drug efflux proteins are associated with the transport of structurally diverse compounds and account for a range of resistance to toxic compounds in microorganisms.
Parkinson et al [72] studied susceptible and resistant isolates of C. glabrata and observed that although there was no change in sterol biosynthesis between these two isolates, the resistant isolate accumulated less fluconazole than the susceptible one and the reduced ability of the resistant strain to accumulate fluconazole was a consequence of energy-dependent drug efflux. Clark et al [73] showed that resistant isolates accumulated less fluorescent dye rhodamine 123 (Rh123) than susceptible cells did. Furthermore, active efflux of Rh123 was observed in azole-resistant isolates of C. albicans and C. glabrata, consistent with the activity of an multidrug-resistant MDR transporter. The efflux mechanism associated with movement of Rh123 appears to play a role in azole resistance inC. glabrata but not in C. albicans, suggesting that azole resistance in C. albicans may be mediated by an alternative efflux pump [74].
Sanglard et al in there study of resistant C. albicans isolates obtained from five AIDS patients observed decreased accumulation of fluconazole associated with up to a 10-fold increase in the mRNA levels of the CDR1 gene in some resistant strains while other resistant isolates overexpressed mRNA from the gene encoding BENr (CaMDR1) and had normal levels of CDR1mRNA [75]. They suggested that CDR1 is involved in the export of several azole derivatives (including fluconazole, itraconazole, and ketoconazole) while BENr confers resistance specifically to fluconazole.
Redding et al in there study of a series of 17 C. albicans isolates cultured from a patient with recurrent episodes (relapses) of oropharyngeal candidiasis observed that patient required progressively higher doses of fluconazole to control the infection after each relapse [76]. The Fluconazole was ineffective after the 14th relapse. Analysis of all isolates by contour-clamped homogeneous electric field electrophoresis confirmed the persistence of the same C. albicans strain throughout all infectious episodes [76]. The sterol content did not differ between susceptible and resistant isolates in this collection, suggesting that the mechanism(s) of resistance does not involve alteration in sterol composition [76]. White examined the expression of several genes of interest in all 17 of these isolates, and found that no expression of ERG1 and ERG7 (genes involved in the ergosterol biosynthetic pathway) was detected [62]. He suggested that high-level azole resistance, at least in this series of isolates, results from the contributions of several mechanisms and prolonged exposure of a strain to one azole may lead to overexpression of genes, such as ERG16and CDR1, that result in cross-resistance to other azoles.
Another emerging source of antifungal resistance is the occurrence of a biofilm, the extracellular matrices produced by microbes themselves which serve to help organisms attach to living or non-viable surfaces [66]. It is estimated that about 65% of all human microbial infections involve biofilms and the majority of invasive diseases produced by C. albicans are associated with biofilm growth [77–79]. It has been demonstrated that drug efflux pumps play a role in the drug resistance of early biofilms [80,81]. In contrast, resistance of mature biofilms does not rely on the known antifungal efflux pumps [80]. It has been hypothesized that a change in membrane sterol composition during biofilm formation might explain resistance to amphotericin B and the azoles [81]. In addition, the MAPK Mkc1p seems to be a regulator of azole resistance in mature biofilms [82].
Resistance to polyene antibiotics is rare, with resistant isolates being confined mostly to the less common species of Candida, such as C. lusitaniae, C. glabrata, and C. guilliermondii [83]. Fryberg suggested that development of resistance occurs by selection of naturally occurring resistant cells, present in small numbers in the population [84]. These naturally resistant cells produce modified sterols that bind nystatin with lower affinity. Athar and Winner, however, have suggested that resistance results from mutation rather than selection [85]. Hamilton-Miller [86] proposed a “biochemical” hypothesis that resistance arises due to changes, either quantitative or qualitative, in the sterol content of the cells. According to this hypothesis, resistant cells with altered sterol content should bind smaller amounts of polyene than do susceptible cells. This decreased binding of polyenes in C. albicans mutants could be attributed to [28] (i) a decrease in the total ergosterol content of the cell, without concomitant changes in sterol composition; (ii) replacement of some or all of the polyene-binding sterols by ones which bind polyene less well, e.g. substitution of ergosterol, cholesterol, or stigmasterol by a 3-hydroxy or 3-oxo sterol [87]; or (iii) reorientation, or masking, of existing ergosterol, so that binding with polyenes is sterically or thermodynamically less favored. Molzahn and Woods [88] reported the isolation and characterization of S. cerevisiae mutants which were resistant to polyenes including nystatin, filipin, and pemaricin. The mutants were allocated to four unlinked genes, pol1, pol2, pol3, and pol5.
Although clinical failure has been observed in patients treated with terbinafine, allylamine resistance in association with clinical use of terbinafine and naftifine has not been found in human pathogenic fungi. However, with the increased use of this agent, resistance may be expected, since Vanden Bossche et al [89] have reported a C. glabrata strain that became resistant to fluconazole and expressed cross-resistance to terbinafine. Sanglard et al reported that CDR1 can use terbinafine as a substrate [90].
Fungi also show adaptive responses to environmental stimuli by activation of several signal transduction pathways in stress conditions [3]. The antifungal drugs induce cellular stress responses needed to overcome its toxic effects, allowing the survival of the fungus [3]. A number of genes involved in adaptation and response to stress and to elucidate the mechanism of action of drugs such as terbinafine, acriflavine, amphotericin B, fluconazole, etc, have been identified [91,92]. Gene expression studies have also contributed to the assessment of the effect of new antifungal agents for T. rubrum, as PHS11A and PH11B recently developed, which act by inhibiting the enzyme fatty acid synthase [93].
CLINICAL IMPLICATIONS, PREVENTION OF ANTIFUNGAL RESISTANCE AND FUTURE DIRECTIONS
The primary factor driving the emergence of antifungal resistance appears to be resulting from the increased use and inappropriate prescribing of systemic antifungal agents [94]. There is no clear evidence as to what dosing strategy should be used during treatment and prophylaxis to best avoid resistance [95]. Ghannoum and Rice [28] suggested measures to avoid and suppress the emergence of antifungal resistance which include (i) prudent use of antifungals, (ii) appropriate dosing with special emphasis on avoiding treatment with low antifungal dosage, (iii) therapy with combinations of existing agents, (iv) treatment with the appropriate antifungal (in cases where the etiological agent is known), and (v) use of surveillance studies to determine the true frequency of antifungal resistance. An increased emphasis on rapid diagnosis of fungi and optimizing therapy according to pharmacokinetic and pharmacodynamic properties and thus reducing exposure to low concentrations of systemic agents should be focussed upon [94,96]. The recent approval of a reference method for the antifungal susceptibility testing of yeast is encouraging and provides a means for performing surveillance studies [97].
Use of combinations of antifungal drugs or use of adjunctive immunostimulatory therapy may be more effective in preventing development of resistance. A variety of immunosuppressive compounds, including cyclosporin and D-octapeptides [98], have been tested and found to counteract antifungal resistance due to efflux pumps. Cernicka, et al. screened a synthetic compound library and identified a chemical that increased the sensitivity of a drug-resistant strain of S. cerevisiae to fluconazole [99]. The compound also increased sensitivity of the pathogenic yeasts Candida albicans and Candida glabrata that expressed efflux pumps [99].
Recently, a team of researchers using detailed genetic, biochemical, and molecular approaches, identified a mechanism controlling multidrug resistance in fungi [100]. They found that yeast induce multidrug resistance via a molecular switch similar to one that removes drugs and other foreign substances from human cells. When the yeast protein Pdr1p binds to anti-fungal drugs or other chemicals, it switches on molecular pumps that remove the drugs from the cell. The research team also showed that this chemical switch also controls drug resistance in an important human pathogenic fungus, Candida glabrata. In humans, a protein called PXR is the drug sensor that turns on genes involved in detoxifying and removing drugs from cells [100]. After binding to drugs, the Pdr1p protein partners with another key mediator of genetic switches called Gal11p. In-depth molecular and structural studies identified the specific area of Gal11p that binds to Pdr1p to induce multidrug resistance [100,101].
A new way to fight drug-resistant fungal infections targeting heat shock protein 90 has been suggested [102]. The Hsp90 chaperone protein provides one mechanism to link temperature with the signalling cascades that regulate morphogenesis, fungal development and virulence. Targeting the molecular chaperone Hsp90 or its downstream effector, the protein phosphatase calcineurin, abrogates resistance to the most widely deployed antifungals, the azoles, which inhibit ergosterol biosynthesis [103]. It was observed that the fungal pathogen Candida albicans is able to resist drug treatment because of an associated molecular chaperone protein called heat shock protein 90 (Hsp90) whose compromising function renders the fungal-fighting drugs known as echinocandins more effective at killing C. albicans laboratory strains and clinical isolates. Hsp90 acted as a kind of thermostat for C. albicans and shutting down the protein’s temperature-sensitivity can shut down the spread of infection. It has been suggested that interfering with Hsp90 function provides a powerful and much-needed strategy to render existing antifungal drugs more effective in the treatment of life-threatening fungal infections [98]. Due to the high degree of conservation in Hsp90, many of the connections in C.albicans may be extrapolated to other fungal pathogens or parasites [98,99].
Photodynamic therapy has been suggested as an alternative treatment for therapy resistant patients, however, the data on this are still limited and in some cases, the aggravation rates are higher than with other methods [104,105,106]. An alternative non-invasive treatment protocol utilizing combinations of VIS-NIR laser beams in association with broadband beams of UV, B and R- LEDs, without usage of photosensitizers, with minimal side effects, for therapy resistant patients suffering from Tinea Pedis, Pityriasis versicolor, or Mycetoma has been demonstrated [107].
CONCLUSION
At this time, antifungal drug resistance is clearly becoming a common problem in patients and is inevitable due to wide availability and use of these agents. There is considerable knowledge concerning the biochemical, genetic and clinical aspects of resistance to antifungal agents. However, sample selection and inadequate information regarding denominators limit current epidemiological data [108,109]. Several variables need to be considered when trying to minimize the risk for development of resistance, including type of drug, intermittent versus continuous dosing during prophylaxis or treatment, the amount of drug administered, the length of treatment, and the immune status of the patient [29]. The availability of molecular genetic tools has led to a rapid expansion in our understanding of the mechanisms by which antifungal resistance emerges and spreads and promises help to develop novel and effective compounds for future use. Research works to study the mechanisms of antifungal resistance, the development of experimental systems in which individual resistance mechanisms can be studied, and establishment of a reproducible method of susceptibility testing will be important components of a strategy to limit the emergence of resistance to these agents and to develop safer and more potent compounds for the future. The current techniques to determine the MIC of a drug include both macro- and microdilution broth methods. However, these remain a time-consuming techniques and quicker methods for the determination of resistance are needed. Targetting efflux pumps, especially ATP binding cassette (ABC) transporters and heat shock protein 90 are the new ways under investigation to fight drug-resistant fungal infections. There is a clear need for the next generation of antifungal agents. New classes of antifungal drugs that are active against azole-resistant isolates, such as the cationic peptide histatin [110] will clearly be important for future treatment strategies [29]. It is likely that the future of antifungal drug therapy lies in drug combinations and improving the immune status of the host. A combination of azoles and cytokines may be an important therapeutic strategy for fungal infections in immunocompromised individuals [111]. Finally, new drug delivery systems may have a place in the treatment of antifungal-drug-resistant infections [29]. As drug resistance continues to develop in pathogenic fungi, ongoing research and developments in the understanding of resistance will find ways to formulate strategies to overcome the resistance.
REFERENCES
1. White TC, Oliver BG, Graser Y, Henn MR, Generating and testing molecular hypotheses in the dermatophytesEukaryot Cell 2008; 7: 1238-45.
2. Rodwell GE, Bayles CL, Towersey L, Aly R, The prevalence of dermatophyte infection in Patients infected with human immunodeficiency virusInt J Dermatol 2008; 47: 339-43.
3. Nalu Teixera de Aguiar Peres An Bras Dermatol2010; 85: 5657-67.
4. Drake LA, Dinehart SM, Farmer ER, Goltz RW, Graham GF, Hardinsky MK, Guidelines of care for superficial mycotic infections of the skin: tinea corporis, tinea cruris, tinea faciei, tinea manuum, and tinea pedisJ Am Acad Dermatol 1996; 34: 282-6.
5. Seebacher C, Bouchara JP, Mignon B, Updates on the epidemiology of dermatophyte infectionsMycopathologia 2008; 166: 335-52.
6. Banerjee M, Ghosh AK, Basak S, Das KD, Gangopadhyay DN, Comparative evaluation of effectivity and safety of topical amorolfine and clotrimazole in the treatment of tinea corporisIndian J Dermatol 2011; 56: 657-62.
7. Kemna ME, Elewski BE, A U.S. epidemiologic survey of superficial fungal diseasesJ Am Acad Dermatol 1996; 35: 539-42.
8. Smith ER, Ferreira JC, Maffei CM, Candido RC, Occurrence of dermatophyte, in nails, feet and hands of university studentsRev Soc Bras Med Trop 2006; 39: 269-71.
9. Godoy-Martinez P, Nunes MR, Tomimori-Yamashita J, Urrutia M, Zaror L, Silva V, Onychomycosis in Sao Paulo, BrazilMycopathologia 2009; 168: 111-6.
10. Bright RS, Lamb RA, Medrano DJ, Rock MF, Miller AJ, Choi CS, Onychomycosis in Ceará (Northeast Brazil): epidemiological and laboratory aspectsMem Inst Oswaldo Cruz 2005; 100: 131-5.
11. Stephenson J, Investigators seeking new ways to stem rising tide of resistant fungiJour Amer Med Assoc 1997; 277: 5-6.
12. Chloe McClanahan, BioFiles2009; 4.3: 10.
13. Wagner DK, Sohnle PG, Cutaneous defenses against dermatophytes and yeastsClin Microbiol Rev 1995; 8: 317-35.
14. Shiraki Y, Ishibashi Y, Hiruma M, Nishikawa A, Ikeda S, Cytokine secretion profiles of human keratinocytes during Trichophyton tonsurans and Arthroderma benhamiae infectionsJ Med Microbiol 2006; 55: 1175-85.
15. Calvin OM, Thomas JL, Fauci Braunwald Kasper Hauser Longo Jameson Loscalzo Eczema, Psoriasis, cutaneous infections, acne and other common skin disordersHarrison’s Principles of Internal Medicine 2008; 1: 17th ed. New York: McGraw-Hill; 318.
16. Liebel F, Lyte P, Garay M, Babad J, Southall MD, Anti-inflammatory and anti-itch activity of sertaconazole nitrateArch Dermatol Res 2006; 298: 191-9.
17. Brooks M, FDA Approves New Topical Antifungal Luliconazole 1%Medscape Medical News Accessed November 18 2013Available at http://www.medscape.com/viewarticle/814470.
18. Brooks M, FDA OKs first topical triazole antifungal for onychomycosis. Medscape Medical News [serial online]. June 102014; Accessed June 16 2014Available at http://www.medscape.com/viewarticle/826480.
19. Vanden Bossche H, Marichal P, Odds FC, Molecular mechanisms of drug resistance in fungiTrends Microbiol 1994; 2: 393-400.
20. Odds FC, Brown AJ, Gow NA, Antifungal agents: mechanisms of actionTrends Microbiol 2003; 11: 272-9.
21. Favre B, Ryder N S, Characterization of squalene epoxidase activity from the dermatophyteTrichophyton rubrum and its inhibition by terbinafine and other antimycotic agentsAntimicrob Agents Chemother 1996; 40: 443-7.
22. Monk JP, Brogden RN, Naftifine. A review of its antimicrobial activity and therapeutic use in superficial dermatomycosesDrugs 1991; 42: 659-72.
23. Ryder NS, Terbinafine: mode of action and properties of the squalene epoxidase inhibitionBr J Dermatol 1992; 39: 2-7.
24. Seyfarth F, Ziemer M, Gräser Y, Elsner P, Hipler UC, Widespread tinea corporis caused by Trichophyton rubrum with non-typical cultural characteristics–diagnosis via PCRMycoses 2007; 50: Suppl 226-30.
25. Lesher JL, JrTinea Corporis Treatment & ManagementLast assessed 30.12.2014 http://emedicine.medscape.com/article/1091473-treatment.
26. Balkis MM, Leidich SD, Mukherjee PK, Ghannoum MA, Mechanisms of fungal resistance: an overviewDrugs 2002; 62: 1025-40.
27. Kauffman CA, Carver PL, Update on echinocandin antifungalsSemin Respir Crit Care Med 2008; 29: 211-9.
28. Ghannoum MA, Rice LB, Antifungal Agents: Mode of Action, Mechanisms of Resistance, and Correlation of These Mechanisms with Bacterial ResistanceClin Microbiol Rev 1999; 12: 501-17.
29. White TC, Marr KA, Bowden RA, Clinical, cellular, and molecular factors that contribute to antifungal drug resistanceClin Microbiol Rev 1998; 11: 382-402.
30. Haria M, Bryson MH, AmorolfineDrugs 1995; 49: 103-20.
31. Polak A, Ryley JF, Mode of action studiesHandbook of Experimental Pharmacology. Chemotherapy of Fungal Diseases 1990; 96: Berlin, Heidelberg: Springer Verlag; 152-82.
32. Muller J, Polak-Wyss A, Melchinger W, Influence of amorolfine on the morphology of Candida albicans and Trichophyton mentagrophytesClin Exp Dermatol 1992; 17: Suppl 118-25.
33. Favre B, Hofbauer B, Hildering KS, Ryder NS, Comparison of in vitro activities of 17 antifungal drugs against a panel of 20 dermatophytes by using a microdilution assayJ Clin Microbiol 2003; 41: 4817-9.
34. Wingfield AB, Fernandez-Obregon AC, Wignall FS, Greer DL, Treatment of tinea imbricata: a randomized clinical trial using griseofulvin, terbinafine, itraconazole and fluconazoleBr J Dermatol 2004; 150: 119-26.
35. da Silva Barros ME, de Assis Santos D, Soares Hamdan J, Antifungal susceptibility testing of Trichophyton rubrum by E-testArch Dermatol Res 2007; 299: 107-9.
36. Chen A, Sobel JD, Emerging azole antifungalsExpert Opin Emerg Drugs 2005; 10: 21-33.
37. Seebacher C, Brasch J, Abeck D, Cornely O, Effendy I, Ginter-Hanselmayer G, OnychomycosisMycoses 2007; 50: 321-7.
38. Stephenson J, Investigators seeking new ways to stem rising tide of resistant fungiJournal of the American Medical Association 1997; 277: 5-6.
39. Marichal P, Vanden Bossche H, Mechanisms of resistance to azole antifungalsActa Biochimica Polonica 1995; 42: 509-16.
40. White TC, Marr KA, Bowden RA, Clinical, cellular and molecular factors that contribute to antifungal drug resistanceClin Microbiol Reviews 1998; 11: 382-402.
41. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard. NCCLS document M27-A1997; Wayne, Pennsylvania: National Committee for Clinical Laboratory Standards;
42. Sanglard D, Odds FC, Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequencesLancet Infect 2002; 2: 73-85.
43. National Committee for Clinical Laboratory Standards1997; NCCLS, Wayne, PA: Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: Approved Standard M27-A;
44. Hudson MM, Antifungal resistance and over-the-counter availability in the UK: a current perspectiveJ Antimicrob Chemother 2001; 48: 345-50.
45. Rex JH, Pfaller MA, Galgiani JN, Bartlett MS, Espinel-Ingroff A, Ghannoum MA, Development of interpretive breakpoints for antifungal susceptibility testing: conceptual framework and analysis of in vitro/in vivo correlation data for fluconazole, itraconazole, and CandidainfectionsClin Infect Dis 1997; 24: 235-47.
46. Ghannoum MA, Rex JH, Galgiani JN, Susceptibility testing of fungi: current status of correlation of in vitro data with clinical outcomeJ Clin Microbioliol 1996; 34: 489-95.
47. Marichal P, Vanden Bossche H, Mechanisms of resistance to azole antifungalsActa Biochimica Polonica 1995; 42: 509-16.
48. Martinez-Rossi NM, Peres ATN, Rossi A, Antifungal resistance mechanisms in dermatophytesMycopathologia 2008; 166: 369-83.
49. Sanglard D, Bille J, Calderone R, Current understanding of the modes of action of and resistance mechanisms to conventional and emerging antifungal agents for treatment of Candida infectionsCandida and candidiasis 2002; USA: ASM Press; 349-83.
50. Parks LW, Casey WM, Prasad R, Ghannoum M, Fungal sterolsLipids of pathogenic fungi 1996; Boca Raton, Fla: CRC Press, Inc; 63-82.
51. Nozawa Y, Morita T, Iwata K, Vanden Bossche H, Molecular mechanisms of antifungal agents associated with membrane ergosterol. Dysfunction of membrane ergosterol and inhibition of ergosterol biosynthesisIn vitro and in vivo evaluation of antifungal agents 1986; B. V. Amsterdam, The Netherlands: Elsevier Science Publishers; 111.
52. Smith KJ, Warnock DW, Kennedy CTC, Johnson EM, opwood V, Van Cutsem J, Azole resistance inCandida albicansJ Med Vet Mycology 1986; 24: 133-144.
53. Orozco A, Higginbotham L, Hitchcock C, Parkinson T, Falconer D, Ibrahim A, Mechanism of fluconazole resistance in Candida kruseiAntimicrob. Agents Chemother 1998; 42: 2645-9.
54. Vanden Bossche H, Willemsens G, Cools W, Marichal HP, Lauwers WFJ, Hypothesis on the molecular basis of the antifungal activity of N-substituted imidazoles and triazolesBiochem. Soc. Trans 1983; 11: 665-7.
55. Vanden Bossche H, Marichal HP, Odds F, Le Jeune L, Coene MC, Characterization of an azole-resistant Candida glabrataisolateAntimicrob. Agents Chemother 1992; 36: 2602-10.
56. Fachin AL, Ferreira-Nozawa MS, Maccheroni W, JrMartinez-Rossi NM, Role of the ABC transporter TruMDR2 in terbinafine, 4-nitroquinoline N-oxide and ethidium bromide susceptibility in Trichophyton rubrumJ Med Microbiol 2006; 55: 1093-9.
57. Cervelatti EP, Fachin AL, Ferreira-Nozawa MS, Martinez-Rossi NM, Molecular cloning and characterization of a novel ABC transporter gene in the human pathogen Trichophyton rubrumMed Mycol 2006; 44: 141-7.
58. Henry KW, Nickels JT, Edlind TD, Upregulation of ERG genes in Candida species by azoles and other sterol biosynthesis inhibitorsAntimicrob Agents Chemother 2000; 44: 2693-700.
59. Chau AS, Mendrick CA, Sabatelli FJ, Loebenberg D, McNicholas PM, Application of real-time quantitative PCR to molecular analysis of Candida albicans strains exhibiting reduced susceptibility to azolesAntimicrob Agents Chemother 2004; 48: 2124-31.
60. Sanguinetti M, Posteraro B, Fiori B, Ranno S, Torelli R, Fadda G, Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistanceAntimicrob Agents Chemother 2004; 49: 668-79.
61. Vermitsky JP, Edlind TD, Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Prd1-like transcription factorAntimicrob Agents Chemother 2004; 48: 3773-81.
62. White TC, Increased mRNA levels of ERG16, CDR, and MDR1correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virusAntimicrob. Agents Chemother 1997; 41: 1482-7.
63. White TC, The presence of an R467K amino acid substitution and loss of allelic variation correlate with an azole-resistant lanosterol 14 a-demethylase in Candida albicansAntimicrob. Agents Chemother 1997; 41: 1488-94.
64. Kelly SL, Lamb DC, Kelly DE, Y132H substitution in Candida albicans sterol 14alpha-demethylase confers fluconazole resistance by preventing binding to haemFEMS Microbiol Lett 1999; 180: 171-5.
65. Kelly SL, Lamb DC, Loeffer J, Einsele H, Kelly DE, The G464S amino acid substitution in Candida albicans sterol 14alphademethylase causes fluconazole resistance in the clinic through reduced affinityBiochem Biophys Res Commun 1999; 262: 174-9.
66. Minnebruggen GV, François IEJA, Cammue BPA, Thevissen K, Vroome V, Borgers M, A General overview on past, present and future antimycoticsThe Open Mycology J 2010; 4: 22-32.
67. Mukherjee PK, Leidich SD, Isham N, Leitner I, Ryder NS, Ghannoum MA, Clinical Trichophyton rubrum strain exhibiting primary resistance to terbinafineAntimicrob Agents Chemother 2003; 7: 82-6.
68. Walsh TJ, Kasai M, Francesconi A, Landsman D, Chanock SJ, New evidence that Candida albicans possesses additional ATP-binding cassette MDR-like genes: implications for antifungal azole resistance 1996J Med Vet Mycol 1996; 35: 133-7.
69. Michaelis S, Berkower C, Sequence comparison of yeast ATP binding cassette (ABC) proteinsCold Spring Harbor Symp Quant Biol 1995; 60: 291-309.
70. Roessner CA, Min C, Hardin SH, Harris HL, McColum JC, Scott AI, Sequence of the Candida albicans erg7 geneGene 1993; 127: 149-50.
71. Iwata K, Drug resistance in human pathogenic fungiEur J Epidemiol 1992; 8: 407-421.
72. Parkinson T, Falconer DJ, Hitchcock C, Fluconazole resistance due to energy-dependent drug efflux in Candida glabrataAntimicrob. Agents Chemother 1995; 39: 1696-9.
73. Clark FS, Parkinson T, Hitchcock CA, Gow NAR, Correlation between rhodamine 123 accumulation and azole sensitivity inCandida species: possible role for drug efflux in drug resistanceAntimicrob. Agents Chemother 1996; 40: 429-25.
74. Lanyi J, Plachy WZ, Kates M, Lipid interactions in membranes of extremely halophilic bacteria. II. Modification of the bilayer structure by squaleneBiochemistry 1974; 13: 4914-20.
75. Sanglard D, Kuchler K, Ischer F, Pagani JL, Monod M, Bille J, Mechanisms of resistance to azole antifungal agents in Candida albicansisolates from AIDS patients involve specific multidrug transportersAntimicrob. Agents Chemother 1995; 39: 2378-86.
76. Redding SJ, Smith J, Farinacci G, Rinaldi M, Fothergill A, Rhine-Chalberg J, Resistance of Candida albicans to fluconazole during treatment of oropharyngeal candidiasis in a patient with AIDS: documentation by in vitro susceptibility testing and DNA subtype analysisClin Infect Dis 1994; 18: 240-2.
77. Donlan RM, Costerton RW, Biofilms: survival mechanisms of clinically relevant microorganismsClin Microbiol Rev 2002; 15: 167-93.
78. Kojic EM, Darouiche RO, Candida infections of medical devicesClin Infect Dis 2004; 17: 255-67.
79. Douglas LJ, Medical importance of biofilms in Candida infectionsRev Iberoam Micol 2002; 19: 139-43.
80. Sanguinetti M, Posteraro B, Fiori B, Ranno S, Torelli R, Fadda G, Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistanceAntimicrob Agents Chemother 2004; 49: 668-79.
81. Mateus C, Crow SA, JrAhearn DG, Adherence of Candida albicans to silicone induces immediate enhanced tolerance to fluconazoleAntimicrob Agents Chemother 2004; 48: 3358-66.
82. Kumamoto CA, A contact-activated kinase signals Candida albicans invasive growth and biofilm developmentProc Natl Acad Sci USA 2005; 102: 5576-81.
83. Martins MD, Rex JH, Resistance to antifungal agents in the critical care setting: problems and perspectivesNew Horizons 1996; 4: 338-44.
84. Fryberg M, Sterol biosynthesis in antibiotic-resistant yeast: nystatinArch. Biochem Biophys 1974; 160: 83-9.
85. Ather MA, Winner HI, Development of resistance by Candidaspecies to polyene antibiotics in vitroJ. Med. Microbiol 1971; 4: 505-17.
86. Hamilton-Miller JMT, Chemistry and biology of the polyene macrolide antibioticsBacteriol Rev 1973; 37: 166-96.
87. Michaelis S, Berkower C, Sequence comparison of yeast ATP binding cassette (ABC) proteinsCold Spring Harbor Symp Quant. Biol 1995; 60: 291-309.
88. Molzahn SW, Woods RA, Polyene resistance and the isolation of sterol mutants in Saccharomyces cerevisiaeJ Gen Microbiol 1972; 72: 339-48.
89. Vanden Bossche H, Marichal HP, Odds F, Le Jeune L, Coene MC, Characterization of an azole-resistant Candida glabrata isolateAntimicrob. Agents Chemother 1992; 36: 2602-10.
90. Sanglard D, Ischer F, Monod M, Bille J, Susceptibility of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitorsAntimicrob. Agents Chemother 1996; 40: 2300-05.
91. Paiao FG, Segato F, Cursino-Santos JR, Peres NT, Martinez-Rossi NM, Analysis of Trichophyton rubrum gene expression in response to cytotoxic drugsFEMS Microbiol Lett 2007; 271: 180-6.
92. Segato F, Nozawa SR, Rossi A, Martinez-Rossi NM, Over-expression of genes coding for proline oxidase, riboflavin kinase, cytochrome c oxidase and an MFS transporter induced by Trichophyton rubrum in AcriflavinMed Mycol 2008; 46: 135-9.
93. Yu L, Zhang W, Liu T, Wang X, Peng J, Li S, Global gene expression of Trichophyton rubrum in response to PH11B, a novel fatty acid synthase inhibitorJ Appl Microbiol 2007; 103: 2346-52.
94. Klepser ME, Ernst EJ, Pfaller MA, Update on antifungal resistanceTrends Microbiol 1997; 5: 372-5.
95. White TC, Marr KA, Bowden RA, Clinical, cellular and molecular factors that contribute to antifungal drug resistanceClinical Microbiology Reviews 1998; 11: 382-402.
96. Maenza JR, Keruly JC, Moore RD, Chaisson RE, Merz WG, Gallant JE, Risk factors for fluconazole resistant Candida in patients with AIDS: prevalence and microbiologyClinical Infectious Diseases 1996; 173: 219-25.
97. Wayne Pa, National Committee for Clinical Laboratory Standards. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard M27-A1997;
98. Monk BC, Goffeau A, Outwitting multidrug resistance to antifungalsScience 2008; 321: 367-8.
99. Cernicka J, Kozovska Z, Hnatova M, Valachovic Hapala I, Riedl Z, Chemosensitisation of drug-resistant and drug-sensitive yeast cells to antifungalsInt J Antimicrob Agents 2007; 29: 170-8.
100. Thakur JK, Arthanari H, Yang F, Pan SJ, Fan X, Breger J, A nuclear receptor-like pathway regulating multidrug resistance in fungiNature 2008; 452: 604-9.
101. Naar AM, Thakur JK, Nuclear receptor-like transcription factors in fungiGenes Dev 2009; 23: 419-32.
102. O’Meara TR, Cowen LE, Hsp90-dependent regulatory circuitry controlling temperature-dependent fungal development and virulenceCell Microbiol 2014; 16: 473-81.
103. Hill JA, Ammar R, Torti D, Nislow C, Cowen LE, Genetic and genomic architecture of the evolution of resistance to antifungal drug combinationsPLoS Genet 2013; 9: e1003390.
104. Lee JW, Kim BJ, Kim MN, Photodynamic Therapy: New Treatment for Recalcitrant Malassezia FolliculitisLasers Surg Med 2010; 42: 192-6.
105. de Oliveira Mima EG, Pavarina AC, Dovigo LN, Vergani CE, de Souza Costa CA, Kurachi C, Susceptibility of Candida Albicans to Photodynamic Therapy in a Murine Model of Oral Can- didosisOral Surg, Oral Med, Oral Pathol, Oral Radiol Endodontol 2010; 109: 392-401.
106. Arits AHM, Mosterd K, Nelemans P, Rooij MJ, Krekels GA, Quaedvlieg PJ, FC20 Three Non-Invasive Treatment Options for Super- ficial Basal Cell Carcinoma: Photodynamic Therapy ver- sus Imiquimod Versus 5-Fluorouracil. TTOP-sBCC TrialMelanoma Research 2010; 20: e38.
107. Penjweini R, Mokmeli S, Becker K, Hans-Ulrich D, Saghafi S, Effects of UV-, Visible-, Near-Infrared Beams in Three Therapy Resistance Case Studies of Fungal Skin InfectionsOptics Photonics J 2013; 3: 1-10.
108. Alexander BD, Perfect JR, Antifungal resistance trends towards the year 2000: Implications for therapy and new approachesDrugs 1997; 54: 657-78.
109. Vazquez JA, Sobel JD, Epidemiologic overview of resistance to oral antifungal agents in the immunocompetent host. Opportunistic infection in the immunocompromised hostExcerpta Med Int Congr Ser 1997 1997; 1-11.
110. Tsai H, Bobek LA, Studies of the mechanism of human salivary histatin-5 candidacidal activity with histatin-5 variants and azole-sensitive and -resistant Candida speciesAntimicrob Agents Chemother 1997; 41: 2224-8.
111. Poynton CH, Immune modulation by cytokines in the treatment of opportunistic infectionsCurr Opin Infect Dis 1997; 10: 275-80.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.