Our Dermatol Online. 2014; 5(4): 401-407
DOI:. 10.7241/ourd.20144.101
Date of submission: 06.08.2014 / acceptance: 17.09.2014
Conflicts of interest: None
TYPE D LYMPHOMATOID PAPULOSIS: AN UNCOMMON VARIANT. A CASE REPORT AND REVIEW OF THE LITERATURE
Gladys Alejandra Paguaga1, Orlando Rodas Pernillo2, Helga María Sarti3
1Graduate of Instituto de Dermatología y Cirugía de Piel, INDERMA, Guatemala, Guatemala
2Departamento de Patología del Hospital Roosevelt, Patmed, Guatemala, Guatemala
3Dermatopathology Department, Instituto de Dermatología y Cirugía de Piel, INDERMA, Guatemala, Guatemala
Corresponding author: Dr Gladys Alejandra Paguaga e-mail: gladyspaguaga@hotmail.com
How to cite this article: Paguaga GA, Pernillo OR, Sarti HM. Type D Lymphomatoid Papulosis: An uncommon Variant. A case report and review of the literature. Our Dermatol Online. 2014; 5(4): 401-407.
Abstract
Lymphomatoid papulosis (LyP) is an indolent form of primary cutaneous T-cell lymphoma, currently classified together with primary cutaneous anaplastic large T-cell lymphoma within the spectrum of CD30-positive lymphoproliferative disorders. It is characterized by presenting as a clinically benign but histopathological malignant disease. Clinical features consist in recurrent waxing and waning red papules. Histopathologically, there are 4 variants recognized, Type A or Hystiocitic type, being the most frecuent of all, Type B or Mycosis fungoides-like, Type C or Anaplastic large-cell lymphoma-like and Type D, the most recently described and uncommon variant with features similar to Cutaneous Aggressive CD8-Positive Cytotoxic T-Cell Lymphoma. We present a case of a 22-year-old female with multiple papules and nodules in trunk and limbs that after histopathological and immunochemical examination was compatible with Type D LyP. It is important to report this case, as a perfect example of an uncommon variant of LyP, with emphasis in its typical clinical, histopathological and inmunohistochemical findings and review of the literature.
Key words: type D lymphomatoid papulosis; CD30-positive lymphoproliferative disorder; CD8-positive lymphoproliferative disorder
Introduction
Lymphomatoid papulosis (LyP) is considered a cutaneous lymphoid dyscrasia because it manifests a paradoxically indolent clinical course despite cytomorphologic, phenotypic, and molecular features overlapping those of lymphoma. Nevertheless, LyP is recognized as a neoplastic process lying at the benign end of the spectrum of CD30+ lymphoproliferative diseases [1]. Clinically it is defined as a rhythmic paradoxical chronic, recurrent, self-healing eruption of erythematous papules and small nodules, characterized by a waxing and waning course and by histopathologic features of cutaneous T-cell lymphoma [2,3]. Four histologic subtypes of LyP are well recognized: (1) type A, characterized by a mixed infiltrate containing large atypical CD30+ cells admixed with small lymphocytes, histiocytes, neutrophils, and/or eosinophils; (2) type B, with a mycosis fungoides (MF)–like histologic picture; (3) type C, characterized by a more monotonous population of large CD30+ cells, similar to those seen in anaplastic large T-cell lymphoma and (4) type D, the most recently described variant, simulating an aggressive epidermotropic CD8-positive T-cell lymphoma. Unlike a true aggressive epidermotropic CD8-positive T-cell lymphoma type D LyP have a similar clinical presentation and an indolent course as the other variants [4]. We describe a case of a 22-year old female patient with the newly described type D LyP who presented typical clinical aspects of LyP but unusual histopathologic and inmunohistochemical features with predominant epidermothropism showing CD8 expression on the immunophenotyping study, a phenotype not seen in the three other common variants.
Case Report
A 22-year-old otherwise healthy female presented with history of multiple self-healing red lesions in trunk and extremities for one month duration. These lesions were associated with mild pruritus. Systemic symptoms were not present. At physical examination we found a polymorphic dermatosis with multiple red papules and nodules, ranging from 0.5 to 2 cm. Some lesions presented central ulceration and others necrotic or serous crusts (Figs 1a – c). The dermatoscopical examination demonstrated thrombosed vessels and serous crusts (Figs 2a – b). Histological examination of a punch biopsy revealed an interesting picture of LyP. The epidermis showed acanthosis and epidermotropism with exocytosis of small lymphocytes. The dermis presented a lymphoid infiltrate in a perivascular and diffuse pattern with cells showing scant cytoplasma and small nuclei with fine cromatine and no atypia. In deep dermis we founded groups of atypical pleomorphic cells with large hypercromatic nuclei arranged in clusters. A few number of atypical mitosis were also seen (Figs 3a – c). A complete inmuhistochemical panel revealed that the malignant infiltrate in deep dermis composed of atypical pleomorphic cells was strongly positive for CD30 (Fig 3d) and partially positive for CD45, but negative for CD3, CD20, anaplastic lymphoma kinase (ALK), epithelial membrane antigen (EMA) and citokeratin. Interestingly, the small T-cells with epidermotropism were strongly positive for CD8 (Fig. 3e) and negative for CD4 and CD3. A complete blood count, urinalysis, HIV, prothrombin and partial thromboplastin time were all within normal limits. With the overall workup of the case we finally concluded that the patient have a newly described type of LyP, known as type D.
 Figures 1A – C. Multiple erythematous papules and nodules in trunk and limbs some ulcerated with necrotic eschar or crust.
|
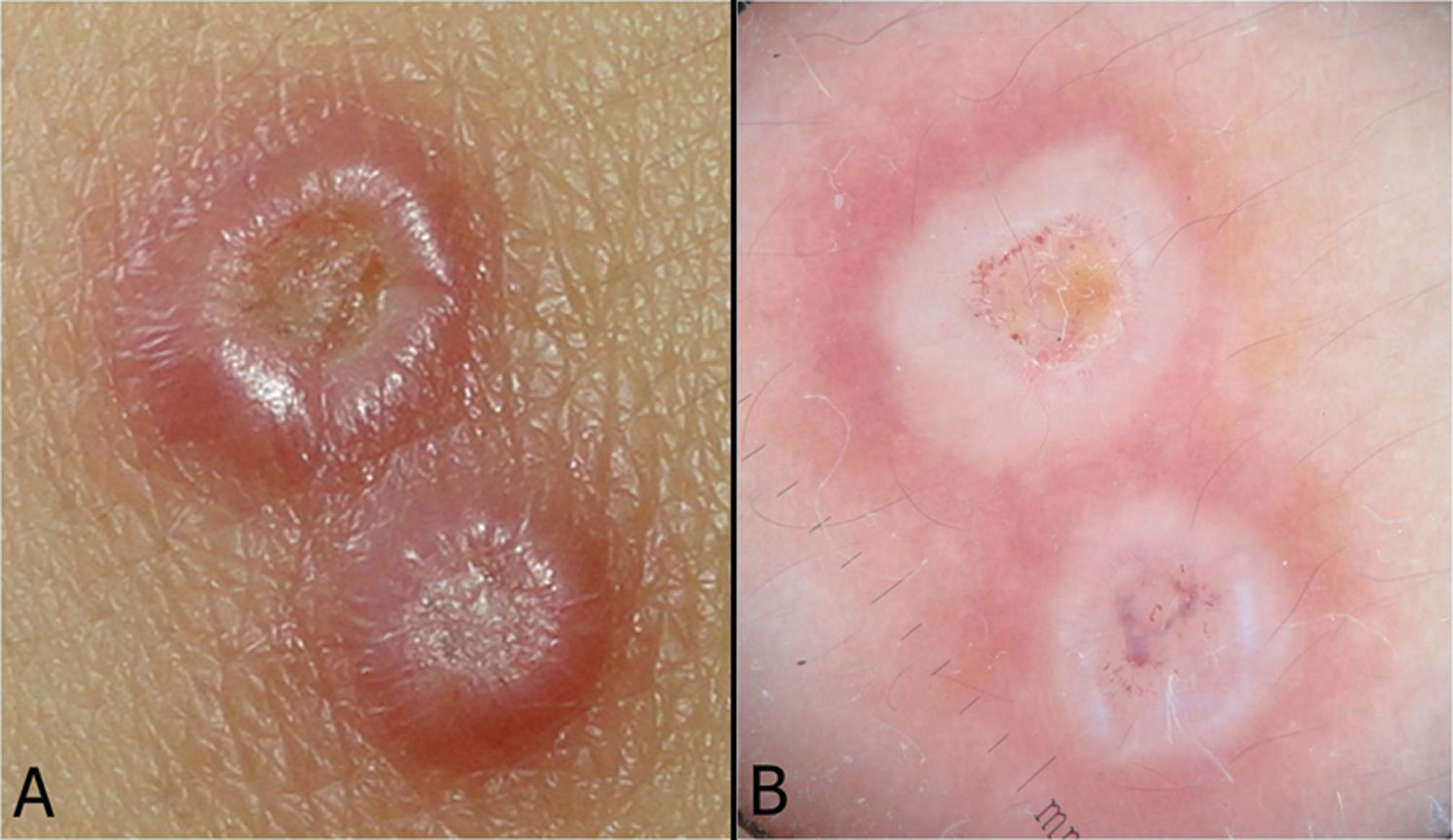 Figures 2A and B. Dermatoscopical features.
|
 Figures 3. (A). Panoramic view. H&E, 4x. (B). Large atypical pleomorphic cells with hyperchromatic nuclei. H&E 40x. (C). Small uniform lymphoid cells wih epidermothropism. H&E 40x
|
 Figures 3D. CD30-positive large atypical pleomorphic cells with hyperchromatic nuclei. 40x
|
 Figures 3E. CD8-positive small uniform lymphoid cells with epidermothropism. 40x
|
Discussion
The term lymphomatoid papulosis originally was used by Macaulay in 1968 to describe „a self-healing rhythmical paradoxical eruption, histologically malignant but clinically benign” [5-7]. Before introduction of the term lymphomatoid papulosis, several cases of continuing self-healing eruptions were diagnosed by some authors as a variety of Mucha-Habermann Disease [6]. Even a few decades ago controversy still surrounded this condition. However, the classification system for cutaneous lymphomas has evolved rapidly, and, during consensus meetings in 2003-2004, the World Health Organization—European Organization for Research and Treatment of Cancer (WHO-EORTC) classification grouped lymphomatoid papulosis among the indolent cutaneous T-cell lymphomas within the spectrum of CD30-positive lymphoproliferative disorders. The rationale for classifying lymphomatoid papulosis as a cutaneous lymphoma is its association with other malignant lymphoproliferative disorders; however, even today some experts hesitate to classify this chronic skin disease as a true malignancy because of its spontaneous resolution and benign clinical course considering it as a pseudolymphomatous inflammatory process [1,7,8]. It was not until recently that there were just three histological variants of LyP known (A, B and C). In 2010, Saggini et al described 9 cases of a newly variant simulating an aggressive epidermotropic CD8-positive T-cell lymphoma that became to be known as type D LyP [2,4]. Epidemiology. The CD30+ cutaneous lymphoproliferative disorders account for approximately 25% of cutaneous T-cell lymphoma cases. The prevalence of lymphomatoid papulosis is estimated to be 1.2-1.9 cases per million population [9]. Although LyP occur at all ages, the peak incidence is between the fourth and fifth decades of life, with a median age of 45 years. It has a slightly predominance in males 1.5:1, even though some few studies have reported a female predominance. Black persons may be less affected by LyP than other racial groups [10-13]. LyP rarely presents in childhood, but when it does it present with age of onset of 12 years. The most common histopathologic subtype described for adult and pediatric-onset is type A LyP [8]. Etiology and pathogenesis. Even though in the past, LyP was considered a pseudolymphomatous disorder, nowadays genetic rearrangement studies have demonstrated that it is clearly an authentic cutaneous lymphoma of low grade malignancy, which together with primary cutaneous anaplastic large cell lymphoma (PCALCL) and borderline CD30-positive lesions is included as a part of a spectrum of CD30-positive cutaneous lymphoproliferative disorders.Borderline CD30 lesions represent cases where histologic features are LyP-like, but clinically behave as lymphoma or cases where histologic features are consistent with PCALCL, but clinically behave as LyP [13,14]. The CD30 antigen is a type I transmembrane glycoprotein with an extracellular domain homologous to tumor necrosis factor and nerve growth factor receptor family members. CD30 is commonly expressed on activated B and T cells [12]. In addition to the CD30+ lymphoproliferative diseases, malignant lymphomas such as Hodgkin disease (HD), node-based systemic anaplastic large cell lymphoma (ALCL), and mycosis fungoides (MF) with large cell transformation may express the CD30 antigen [9,13]. The etiology of LyP remains uncertain [14,15]. Some investigators hypothesized that a retrovirus related to Human T lymphotropic virus-1 may be responsible for the activation and clonal expansion of the LyP cells. Such a virus can be suspected because of the usual adult onset of the disease, cutaneous lesions, and the presence of large, atypical T cells resembling the transformed cells of adult T-cell leukemia/lymphoma. Further research is needed to prove this hypothesis [12]. In the other hand, CD30 signaling is known to have an effect on the growth and survival of lymphoid cells, and one hypothesis is that genetic instability and accumulated genetic defects may have a role in the development of lymphomatoid papulosis and the progression to associated neoplasms [9]. Clinical findings. It presents as a recurrent polymorphic cutaneous eruption characterized by generalized self-healing (within a period of 20 days to 2 months) erythematous asymptomatic to mildly pruritic crops of waxing and waning papules and nodules that are at different developmental stages and progress in recurrent episodes. Some lesions ulcerate and develop a necrotic eschar or crust. Their size varies, but usually do not exceed 2cm [7-10,14-16]. They can be a few or a thousand of them, being in mayority of cases scattered and symmetrically distributed affecting principally trunk and proximal limbs. Mucosal membranes are usually not affected, but there are cases of involvement oral and vulvar mucosa. Unless accompanied by systemic lymphoma, most patients have no constitutional symptoms. Unusual presentations described include localized forms being more frequently seen in children and young adults, pustulous variants, LyP variants affecting the mucosal membranes and hidroa-vacciniforme-like variants [9,11,14,16,17-20]. Lesions heals spontaneously within 2-8 weeks, leaving a hypopigmented or hyperpigmented, depressed, oval, and varioliform scar, especially if the previous lesion was an ulcero-necrotic nodule [9,11,14,21]. Evolving lesions have been described under dermoscopy. The initial papular lesion shows a vascular pattern of tortuous vessels radiating from the center. A white structureless area is seen around the vessels. More mature lesions, hyperkeratotic papules, looked similar except the vascular pattern in the center of the lesion is darker. As the lesions progress to necrotic ulcerations, the vascular pattern is only seen at the periphery, while the center of the lesions presents brownish-gray areas. The final, or cicatricial phase, is similar except no vessel pattern is seen [9]. Histopathological findings. The tipical lesions of LyP present as a wedge infiltrate with a varied number of large atypical cells that can be solitary or clustered [1,11]. Since 2010, there are 4 histological types of LyP well described [2,4,11]. The most common histopathologic subtype of LyP is Type A or hystiocitic type, it represents the prototypic subtype of LyP originally described by Macaulay. It is characterized by a mixed infiltrate containing large, atypical, CD30-positive lymphocytes with bizarre-shaped nuclei, resembling Reed-Sternberg cells from Hodgkin’s lymphoma presented in a wedge-shaped distribution throughout the dermis mixed with various numbers of inflammatory cells (small lymphocytes histiocytes, neutrophils and/or eosinophils) and no epidermothropism. Type B or Mycosis Fungoides (MF)-like is less common and is characterized by small, atypical, CD30 lymphocytic cells with hyperchromatic cerebriform nuclei (resembling the lymphocytes known as Sézary cells found in mycosis fungoides) that are distributed in a bandlike pattern, with concomitant epidermotropism (similar to that seen in patch/plaque stage of mycosis fungoides). In addition, type B is probably the most ambiguous histopathologic variant of LyP, as besides the similarities to MF and lack of large anaplastic cells, which are the features that distinguish it from the conventional (type A) variant of LyP, in many cases reported in the past, neoplastic cells lacked CD30 expression too, thus being a source of conceptual confusion and of diagnostic problems. Type C or Anaplastic large-cell lymphoma-like is rarer and consists of a monotonous population of large, atypical, CD30-positive cells diffusely infiltrating the dermis, with fewer associated inflammatory cells than those seen in other types. Type D, simulating an aggressive epidermotropic CD8-positive T-cell lymphoma is characterized by uniform small to medium-sized, CD8-positive lymphocytic cells with pagetoid reticulosis-like epidermothropism and large, atypical, pleomorphic CD30-positive cells distributed in clusters throughout the dermis. Other important features are absence of eosinophils and neutrophils and common vasculitic changes possibly reflective of the concomitant cytokine milieu [1-4,11,22-25]. Inmunohistochemical findings. LyP is characterized immunohistochemically by the presence of large atypical neoplastic cells that present phenotype of activated T- helper cells that express typically CD4+, CD30+ and CD25+. Half of the cases, regardless of the subtype are CD56+. Even though this marker is associated to a poor prognosis in others lymphoproliferative disorders, this is not the case in LyP. Cytotoxic molecules that do not imply a different clinical behavior as Granzime B and Perforin may also be found. In the case of type D LyP, the hallmark feature is the presence of small T-cells with epidermothropism that express CD4+ in addition to the CD30+ neoplastic cells [3,11,26]. Differential diagnosis. LyP should be differentiated first from other conditions that present atypical large cell infiltrate. In the past it was considered that the presence of the CD30 antigen was exclusive of some lymphomas, but nowadays CD30+ has being confirmed in various benign cutaneous conditions. The differential diagnosis of LyP type A from cutaneous CD30-positive anaplastic large cell lymphoma (ALCL) and Hodgkin Disease (HD) can be difficult on both clinical and morphologic grounds. Lymphomatoid papulosis, cutaneous CD30-positive ALCL, and HD belong to the spectrum of primary cutaneous CD30+ lymphoproliferative disorders,and some cases may have similar clinical presentation and histopathologic findings; however, the presence of self-healing papules and nodules is more characteristic of LyP.In LyP type A the size of the papules and nodules usually does not exceed 2cm, whereas in primary cutaneous CD30-positive ALCL and HD the tumors are usually larger than 2cm and persistent. Morphologically, the anaplastic large cells and RS-like cells that characterized LyP type A may resemble the neoplastic cells that are present in CD30-positive ALCL and HD. The differential diagnosis of LyP from CD30-positive ALCL is based on the number of large, anaplastic, and Reed Sternberg-like cells. In CD30-positive ALCL the atypical cells represent the majority of the cellular infiltrate, whereas in LyP type A the infiltrate contains mostly small, mature lymphocytes and only occasional large, atypical cells. Immunophenotypically, the large, atypical cells of LyP and CD30-positive ALCL have an identical profile: they are positive for CD30 and CD45 and are negative for CD15. The differential diagnosis of LyP type A from HD is based almost exclusively on the immunophenotypic findings: in contrast to LyP, the RS cells of HD are negative for CD45 and positive for CD15 [15,26]. The cases negative for CD30 (LyP type B) should be distinguished from the papular variant of mycosis fungoides. Type C LyP could be difficult to distinguished from anaplastic large cell lymphoma (ALCL), because many times they exhibit clinical, histological and immunochemical overlap [11,24,26]. The differential diagnosis of Type D LyP includes primary cutaneous aggressive epidermotropic CD8? T-cell lymphoma (CTCL), mycosis fungoide, pityriasis lichenoides et varioliformis acuta (PLEVA) and pagetoid reticulosis. The cornerstone for distinguishing between these disease entities is the clinic-pathologic correlation. Ultimately, it is the indolent waxing and waning clinical behavior characteristic of LyP that permits this critical distinction to be made. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma (CTCL) unlike LyP is characterized by the rapid onset of patches, plaques, nodules, and tumors frequently exhibiting necrosis and ulceration. The clinical course is aggressive with a median survival of 32 months. Extracutaneous spread to unusual sites including the testes, lung, and central nervous system is a frequent feature. Histologically, it shows characteristically striking epidermotropism of atypical lymphocytes with a CD8+ cytotoxic phenotype, hence bearing many similarities to our case. Most importantly, CD30 is usually negative. A papular variant of MF has been described recently; however, prominent (pagetoid reticulosis-like) epidermotropism is not a feature of this variant of MF, and lesions are stable without the typical spontaneous resolution observed in LyP. In addition, neoplastic lymphocytes in papular MF have always been reported as being CD4+, CD8-, and CD30-. A more difficult differential diagnosis is with PLEVA, as this disorder shares some clinical and histopathologic features with LyP, and most reported cases had a CD8+phenotype. In addition, monoclonal rearrangement of the TCR genes has been observed in a variable proportion (10% to 100%) of cases of PLEVA studied in the past. Finally, cases reported as ‘‘clonal CD30+ PLEVA’’ have further contributed to the existing confusion in this field of dermatology and dermatopathology. It is quite plausible that at least some among the cases reported in the past as conventional or febrile ulcero-necrotic PLEVA with ‘‘atypical’’ and/or CD30+ lymphocytes may in fact be better classified as the peculiar variant of LyP that we described in this article. In contrast, epidermotropism with a pagetoid reticulosis-like pattern is not a typical feature of PLEVA, and presence of medium sized, atypical, pleomorphic lymphocytes should be considered as virtually ruling out this diagnosis. Another differentiating feature is the absence of necrotic keratinocytes in LyP, in contrast to the apoptotic cells commonly found in PLEVA. Pagetoid Reticulosis PR (Woringer–Kolopp disease) was also considered based on the histologic findings. It is regarded as a variant of MF in the current classifications. The clinical setting is rather different from LyP, manifesting as solitary or localized scaly or hyperkeratotic plaques with slow growth and indolent behavior, typically involving the extremities. Histologically, it is characterized by striking epidermotropism by atypical pagetoid cells, and the phenotype is frequently CD8+. Unlike LyP, the infiltrate tends to be more superficial and not wedge shaped, and tumor cells tend to be almost exclusively confined to the epidermis. Interestingly, CD30 positivity has also been increasingly reported in PR, and so confident distinction from this variant of LyP based on the immunophenotype alone is not reliable and ultimately depends on clinicopathologic correlation [1-4]. Clinical Course and Prognosis. Patients with LyP have a chronic, indolent, self-healing, recurrent and relapsing clinical course regardless of treatment modalities [9,11]. Spontaneous regression of LyP is seen almost universally and recurrence in crops establish a chronicity that generally last for years, even though most of patients with LyP remain in good health. No clinical or pathological features can predict increased risk for developing malignancy and although it is not an aggressive malignant process, patients with LyP have an increased risk for developing a nonlymphoid tumour or more commonly a lymphoma (10-20%) including mycosis fungoides, Hodgkin’s disease, and cutaneous and systemic CD30+ large-cell lymphoma and 10% of these are associated with extracutaneous involvement [3,9,21,22,27-30]. The prognosis of LyP is characterized by disease-specific 5-year survival rates around 100%. Especially in Type D LyP, this is clearly different from the aggressive clinical course and poor prognosis of other lymphomas that are included in the differential diagnosis of this new variant, particularly the aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma [4]. Treatment. The treatment options proposed are varied, however none of them have proven to be completely effective, causing relapses soon after treatment is suspended. While some authors proposed aggressive therapies based on the increased risk of developing another lymphoproliferative disorder, others disagree based in the concept that treatment modalities wont modified the natural history of LyP [11,31]. Treatment should be individualized. It goes from simple observation to the use of drugs when this is required [22]. Cases with few lesions that resolve without scars do not require active treatment [31]. Patients with LyP need to be educated about their disease. They should be told of its benign clinical nature and that it is not lymphoma. On the other hand, they need to realize that their disease can evolve into lymphoma and that the exact risk is still unknown. It is very important to inform about the possible changes that might indicate malignancy: persistence of nodules, increased size of lesions, B symptoms, and lymph node enlargement. After a history and physical examination, patients should have the following baseline investigations: complete blood cell count, liver function tests, a roentgenogram of the chest, and a computed tomographic scan of the abdomen.A bone marrow examination is indicated if the complete blood cell count is abnormal. These investigations can serve as baseline in the event of change and negative results can help reassure the often concerned patient that there is no evidence of lymphoma. Because there is an acumulative risk of transformation into lymphoma these patients need to be examined regularly. At least, an annual examination is indicated, or more frequently depending on the severity of the disease and the type of treatment chosen [32]. Overall is difficult to assess efficacy of the different treatment options because LyP is a self-healing disorder and even though there is no curative treatment, the use of topical steroids, systemic steroids, PUVA, retinoids and methotrexate may induce temporary remissions. The used of low dose oral methotrexate (15-20 mg per week) alone or with PUVA have been reported to induce the best results, causing long remissions after low dose treatments. It has proven safe and effective even in severe cases in which it is well tolerated. A few reports also have found that topical carmustine, topical nitrogen mustard, topical MTX, topical imiquimod cream, intralesional interferon, low-dose cyclophosphamide, chlorambucil, excimer laser therapy, photodynamic therapy, antibiotics as tetracycline, antiviral as acyclovir and dapsone help disease suppression. Surgical excision in cases with few lesions is also reported. Polychemotherapy is not indicated. Overall, treatment in each case should be individualized and risk/benefit ratio of these therapies should be carefully assessed [9,10,14,21,31,33].
Conclusion
The diagnosis of LyP can be quite challenging due to the specific histologic features of the disease making misdiagnosing more likely. Moreover, unusual variants like Type D LyP may pose a significant diagnostic problem, especially if immunochemical examination is not available. In this article we describe a case of a newly LyP variant know as Type D in a 22-year old female. We emphasized the importance of the overall workup made; with clinical, dermatoscopical, histological and immunochemical features that support the diagnosis. Since its original description in 2010 until now this recently described variant has rarely been reported so we hope that with our case we can help others get to know this uncommon variant. As others type D LyP cases our patient presented typical clinical aspects of LyP but with unusual histopathologic and inmunohistochemical features, with predominant epidermothropism with CD8 expression on the immunophenotyping study, a phenotype not seen in the other three more common variants. These findings resembled primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma, a lymphoma that is characterized by an aggressive course with very poor prognosis. With that said, we believe that recognition of this variant allows correct classification of these cases, since the differentiation of LyP from other lymphoproliferative disorders is crucial for proper management of the patients.
REFERENCES
1. Magro C, Crowson N, Morrison C, Merati K, Porcu P, Wright E. CD8+ lymphomatoid papulosis and its differential diagnosis. Am J Clin Pathol. 2006;125:490-501.
2. Saggini A, Gulia A, Argenyi Z, Fink-Puches R, Lissia A, Magaña M, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic t-cell lymphoma. Description of 9 cases. Am J Surg Pathol. 2010;34:1168–75.
3. Sim J, Kim Y. CD8+ lymphomatoid papulosis. Ann Dermatol. 2011;23:104-7.
4. Cardoso J, Duhra P, Thway Yi, Calonje E. Lymphomatoid papulosis type D: A newly described variant easily confused with cutaneous aggressive CD8-positive cytotoxic t-cell lymphoma. Am J Dermatopathol. 2012;34:762–5.
5. Macaulay W. Lymphomatoid papulosisA continuing self-healing eruption, clinically benign—histologically malignant. Arch Dermatol. 1968;97:23-30.
6. Macaulay W. Lymphomatoid papulosis. Int J Dermatol. 1978;17:204-12.
7. Souza A, Azhary R, Camilleri M, Wada D, Appert D, Gibson L. In search of prognostic indicators for lymphomatoid papulosis: A retrospective study of 123 patients. J Am Acad Dermatol. 2012;66:928-37.
8. Souza A, Camilleri M, Wada D, Appert D, Gibson L, Azhary R. Clinical, histopathologic, and immunophenotypic features of lymphomatoid papulosis with CD8 predominance in 14 pediatric patients. J Am Acad Dermatol. 2009;61:993-1000.
9. Zic J. Lymphomatoid papulosis. emedicine.medscape.com; Updated: Jan 24, 2012 http://emedicine.medscape.com/article/10989. 54-overview
10. Sanches J, Zavaloni C, Festa Neto C. Lymphoproliferative processes of the skin. Part 2 – Cutaneous T-cell and NK-cell lymphomas. An Bras Dermatol. 2006;81:7-25.
11. Alperovich M. Lymphomatoid papulosis. Dermatol Argent. 2011;17: 354-64.
12. Ishiji T, Takagi Y, Niimura M. Cutaneous lymphomas in Tokyo: analysis of 62 cases in a dermatology clinic. Int J Dermatol. 2001;40:37-40.
13. Liu H, Hoppe R, Kohler S, Harvell J, Reddy S, Kim Y. CD30+ cutaneous lymphoproliferative disorders: The Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-58.
14. Rodríguez Peralto J, Calzado L, Vanaclocha F. En: Herrera Ceballos E, Moreno Carazo A, Requena Caballero L, Rodríguez Peralto J. editores. Dermatología: correlación clínico-patológica. Barcelona: Signament edicions, S.L., 2007;pp:447-50 .
15. Sioutos N, Asvesti C, Sivridis E, Aygerinou G, Tsega A, Zakopoulou N, Zographakis I. Lymphomatoid papulosis type A: clinical, morphologic, and immunophenotypic study. Int J Dermatol. 1997;36:514-7.
16. Hsu Y, Su L, Hsu Y, Tsai T, Hsiao C. Localized lymphomatoid papulosis. J Am Acad Dermatol. 2010;62:353-5.
17. Eminger L, Shinohara M, Kim E, Heymann W. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531–4.
18. Yancovitz M, Walters R, Kamino H, Brown L. Acral lymphomatoid papulosis. J Am Acad Dermatol. 2010;62:530-1.
19. Chimenti S, Fargnoli M, Pacifico A, Peris K. Mucosal involvement in a patient with lymphomatoid papulosis. J Am Acad Dermatol. 2001;44:339-41.
20. Tabata N, Aiba S, Ichinohazama R, Kikuchi K, Aoyama H, Watanabe M, et al. Hydroa vacciniforme-like lymphomatoid papulosis in a Japanese child: A new subset. J Am Acad Dermatol. 1995;32:378-81.
21. Hurtado J, Bravo F. Papulosis linfomatoide. Dermatol Per. 2000;10:130-2.
22. Lin J, Shiau W, Lu C, Tsai C, Wong W. Chung-Yee Hui R. Lymphomatoid papulosis: a clinical and histopathologic review and follow-up study of 34 cases in Taiwan. Derm Sinica. 2011;29:8-12.
23. Ally M, Robson A. Pagetoid reticulosis in lymphomatoid papulosis type D and mycosis fungoides, mimicking aggressive epidermotropic CD8+ (‘‘Berti’’) lymphoma. J Am Dermatol. 2012;66:AB81.
24. Vonderheid E, Kadin M, Gocke C. Lymphomatoid papulosis followed by pityriasis lichenoides: a common pathogenesis? Am J Dermatopathol. 2011;33:835-40.
25. Wu W, Tsai H. Lymphomatoid papulosis histopathologically simulating angiocentric and cytotoxic t-cell lymphoma. Am J Dermatopathol. 2004;26:133-5.
26. Flann S, Orchard G, Wain M, Russell-Jones R. Three cases of lymphomatoid papulosis with a CD561 immunophenotype. J Am Acad Dermatol. 2006;55:903-6.
27. Louvet S, Martin A, Troussard X, Galateau E, Moreau A, Reman O, et al. Spectrum of CD30 lymphoproliferative diseases from lymphomatoid papulosis to anaplastic large cell lymphoma. Int J Dermatol. 1996;35:842-8.
28. Heald P, Subtil A, Breneman D, Wilson L. Persistent agmination of lymphomatoid papulosis: An equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2007;57:1005-11.
29. Siddiqui M, Sullivan S, Al-Mofadhi A. Lymphomatoid papulosis and FK 506. Int J Dermatol. 1997,36:198-212.
30. Tokuriki A, Kiyohara T, Ido T, Kumakiri M. A case of lymphomatoid papulosis with extensive limb disease followed by extracutaneous involvement and acquired ichthyosis. Acta Derm Venereol. 2012;92:278–9.
31. Narro R, Lacy R, Hojyo M. Papulosis linfomatoide. Reporte de dos casos tratados con PUVA. Dermatología Rev Mex. 2007;51:158-60.
32. Demierre M, Goldberg L, Kadin M, Koh H. Is it lymphoma or lymphomatoid papulosis? J Am Acad Dennatol. 1997;36:765-72.
33. Bergstrom J, Jaworsky C. Topical methotrexate for lymphomatoid papulosis. J Am Acad Dermatol. 2003;49:937-9.
Comments are closed.