Our Dermatol Online. 2011; 5(1): 65-67
DOI:. 10.7241/ourd.20141.17
Date of submission: 17.09.2013 / acceptance: 16.10.2013
Conflicts of interest: None
LESCH-NYHAN SYNDROME: A RARE DISORDER OF SELF-MUTILATING BEHAVIOR
Mrinal Gupta, Vikram K. Mahajan, Vikas Sharma, Pushpinder S. Chauhan, Karaninder S. Mehta
Department of Dermatology, Venereology and Leprosy, Dr. R. P. Govt. Medical College, Kangra (Tanda)-176001 (Himachal Pradesh), India
Corresponding author: Prof. Vikram K. Mahajan e-mail: vkm1@rediffmail.com
How to cite an article: Gupta M, Mahajan VK, Sharma V, Chauhan PS, Mehta KS. Lesch-Nyhan Syndrome: a rare disorder of self-mutilating behavior. Our Dermatol Online. 2014; 5(1): 65-67.
Abstract
This paper describes Lesch-Nyhan syndrome in a 1-year-old boy. This X-linked recessive error of purine metabolism presents in infancy with a constellation of mental and developmental retardation, self-mutilating behavior, neurological features and abnormal urine uric acid: creatinine ratio. The basic defect is deficiency in phosphoribosyl transferase production but exact pathomechanism for clinical symptomatology remains un-elucidated. No specific medical treatment is available.
Key words: Dysarthria; Hyperuricemia; Hypoxanthine-guanine phosphoribosyl transferase; Metabolism, Inborn Errors; Purine metabolism; Self-mutilating behavior
Introduction
Lesch-Nyhan syndrome (LNS) is an extremely rare X-linked recessive error of purine metabolism due to severe inborn deficiency of hypoxanthine-guanine phosphoribosyl transferase (HPRT) enzyme. This enzyme is normally present in every cell but brain (basal ganglia) has the highest concentration and is essential for normal metabolism of hypoxanthine. The HPRT gene is located on the long arm of the X chromosome at Xq26.1 and the complete amino acid sequence for HPRT is approximately 42 kb and split into 9 exons [1]. Clinically, LNS manifests with consistent and compulsive self-biting behavior that usually begins with the eruption of teeth, hyperuricemia, mental retardation, cerebral palsy, dysarthric speech, choreoathetosis initially and spasticity and dystonia later. The self-mutilating behavior persists and results in partial or total destruction of lower lip and/or amputation of fingers, toes, and sometimes of tongue. Most patients do not survive childhood due to renal or respiratory complications. The survival beyond 20 years of age is an exception but the life span may be normal for patients with partial HPRT deficiency (Kelley-Seegmiller syndrome). The estimated prevalence of LNS is 1 in 380,000 live births in Canada and 1 in 235,000 live births in Spain [2] and it has been rarely reported from India.
Case Report
1-year-old male child was brought for dermatological assessment of failure to thrive and lacerations over lower lip, thumbs and left index finger due to self biting. Historically, the child had developed the habit of self biting at 10 months of age roughly coinciding with eruption of teeth. The ulcers would heal spontaneously after bandaging but recur within a day after its removal. He was the only child born to non-consanguineous parents after a normal gestation/pregnancy. He was unable to hold his head yet. He measured 56cm (50th centile 74.4cm) in height, weighed 6kg (50th centile 9.04kg) and had occipito-frontal circumference of 45.5cm (50th centile 46.5cm) suggestive of severe growth retardation. His bone age was consistent with his chronological age and mental age was 8 months. Cutaneous examination (Fig. 1) showed well-defined ulcers/lacerations with crusting and scarring at places involving both the thumbs and left index finger. Nails of the involved fingers were dystrophic. A single, well-defined, deep ulcer with ragged margins and some scarring was present over lower lip. There was no regional lymphadenopathy. Neurological examination showed chorea, hyper-reflexia and positive Babinski’s sign. Other systemics examination and laboratory investigations (blood cell counts, hepatorenal functions, urinalysis and chest radiograph) were normal. The serum uric acid levels were 6.5mg/dl (normal 1.7-5.8 mg/dL) and the urine uric acid: creatinine ratio was 3:4 (normal 2:5-3:5).HPRT enzyme estimation in erythrocyte lysate or skin fibroblasts was not done for lack of availability/affordability. However, features of self-mutilating behavior, physical and mental retardation, neurological features, and abnormal urine uric acid: creatinine ratio were suggestive of LNS.
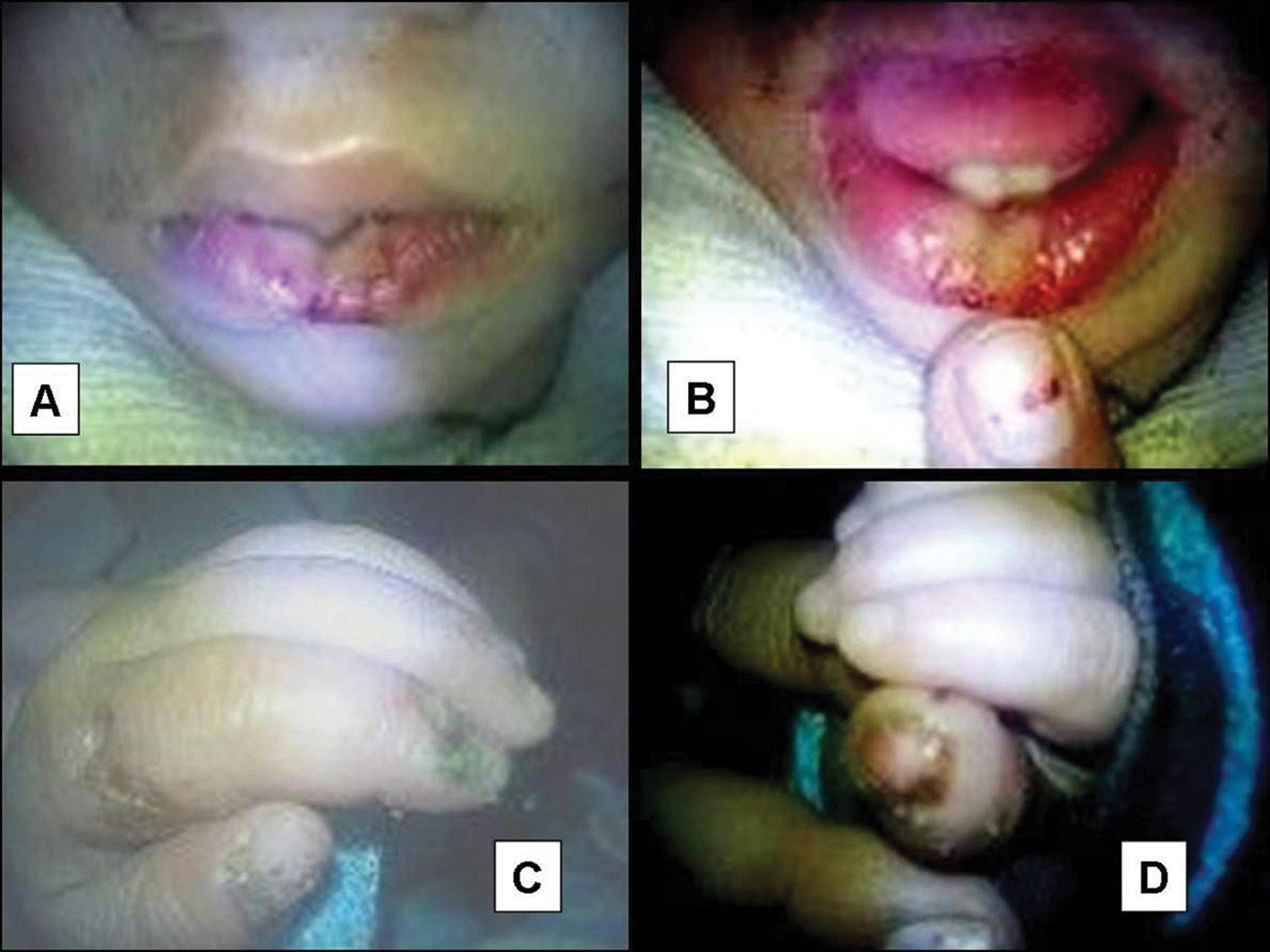 Figure 1. A well defined deep ulcer with ragged margins over lower lip (A & B). Ulcers and lacerations with crusting and scarring over left index finger and thumb (C) and right thumb (D). Note dystrophic nails of the involve digits.
|
Discussion
Lesch-Nyhan syndrome is extremely rare genodermatosis having developmental, behavioral, neurological, and biochemical abnormalities. Born normally, the infant develops growth retardation and neurologic signs after several months. Extrapyramidal signs (chorea, dystonia) and pyramidal tract signs (hyper-reflexia, sustained ankle clonus, positive Babinski sign, scissoring) become evident usually by 8-12 months. There may be moderate to severe mental retardation along with dysarthric speech. The self-injurious behavior usually begins with self-biting at 1 year or may be delayed until teens. Other patterns of self-injurious behavior emerge over time, albeit, aggressive behavior may decline in few after 10-12 years of age. Self-biting is intense and substantial loss of tissue around the lips and partial or total amputation of fingers/toes may result. The biting pattern can be asymmetric, with preferential mutilation of the left or right side of the body [3]. Interestingly, the pain sensations are intact and the child screams in pain on self-biting. The exact mechanism of neurologic and behavioral symptoms in LNS is poorly understood. The metabolism of both hypoxanthine and guanine is affected; guanine triphosphate and adenosine have substantial effects on neural tissues through dopamine and adenosine systems. Dopamine and adenosine systems are also linked through the neuroprotective role of adenosine in preventing neurotoxicity [4]. The majority of classic LNS patients have low or undetectable levels of the HPRT enzyme while partial deficiency in HPRT with more than 1.5-2.0% enzyme (Kelley-Seegmiller syndrome) is associated with hyperuricemia and variable neurologic dysfunction (neurologic HPRT deficiency) [4]. The deficiency of HPRT leads to accumulation of hypoxanthine, particularly in cerebrospinal fluid, excessive uric acid production, and consequent hyperuricemia. However, the behavior disorder is not due to excess hypoxanthine or hyperuricemia. Depression of dopaminergic neurons due to decreased arborization of terminal neurons too has been implicated as the causative factor but needs substantiation [5]. The diagnosis of LNS is mainly clinical and by serum uric acid estimation. Assay for HPRT enzyme in erythrocyte lysate or demonstration of mosaicism in skin fibroblasts will confirm the diagnosis or carrier state but poor availability and high cost often limit their use in clinical practice. Prenatal diagnosis is possible by amniocentesis or chorionic villous sampling. No drug is effective for preventing neurological dysfunction; prevention of self mutilation by using restraints or protective mouth guard, even tooth extraction, remains the mainstay of management [6]. Treatment with diazepam for anxiety, risperidone for aggressive behavior, and carbamazepine or gabapentin for mood stabilization is advocated. Gabapentin was proved more useful in controlling neuropsychiatric symptoms than carbamazepine or sodium valporate [7]. Conventionally, allopurinol has been used for hyperuricemia to prevent renal complications while fabuxostat remains unstudied. Rasburicase, a recombinant urate oxidase enzyme, effectively reduced plasma urate levels and improved kidney function in a neonate with LNS [8]. Behavioral therapy to manage mood swings has been found successful in selective cases. There is not enough evidence that bone marrow transplantation is beneficial as no improvement in neurologic or behavioral symptoms has been observed [9]. Gene therapy wherein DNA cloned from HPRT gene transferred to HPRT deficient cells in vitro appears promising [10].
REFERENCES
1. Kim SH, Moores JC, David D, Respess JG, Jolly DJ, Friedmann T. The organization of human HPRT gene. Nucleic Acid Res. 1986;14:3103-18.
2. Torres RJ, Puig JG. Lesch-Nyhan syndrome. Drugs of the Future. 2010;35:421-27.
3. Robey KL, Reck JF, Glacomini KD, Barabas G, Eddey GE. Modes and patterns of self-mutilation in persons with Lesch-Nyhan disease. Dev Med Child Neurol. 2003,45:167-71.
4. Wilson JM, Young AB, Kelley WN. Hypoxanthine-guanine phosphoribosyl transferase deficiency – the molecular basis of the clinical syndromes. N Eng J Med. 1982;305:900-8.
5. Lloyd KG, Hornykiewicz O, Davidson L, Shannak K, Farley I, Goldstein M, et al. Biochemical evidence of dysfunction of brain neurotransmitters in the Lesch-Nyhan syndrome. N Eng J Med. 1981;305:1106-11.
6. Hall S, Oliver C, Murphy G. Self-injurious behavior in young children with Lesch-Nyhan syndrome. Dev Med Child Neurol. 2001;43:745-9.
7. Rasul CH, Mostafa KG, Nasrin E. Role of antiepileptics in Lesch-Nyhan syndrome. Glob Adv Res J Med Med Sci 2012;2:001-004.
8. Nyhan WL, Parkman R, Page T, Gruber HE, Pyati J, Jolly D, et al. Bone marrow transplantation in Lesch-Nyhan disease. Adv Exp Med Bio. 1986;195:167-70.
9. Roche A, Pérez-Dueñas B, Camacho JA, Torres RJ, Puig JG, Garcia-Cazorla A, et al. Efficacy of Rasburicase in hyperuricemia secondary to Lesch-Nyhan syndrome. Am J Kidney Dis. 2009;53:677-80.
10. Urbach A, Schuldiner M, Benvenisty N. Modeling for Lesch-Nyhan disease by gene targeting in human embryonic stem cells. Stem Cells. 2004;22:635-41.
Comments are closed.