DOI: 10.7241/ourd.20124.70 article in PDF
Our Dermatol Online. 2012; 3(4): 321-325
Date of submission: 08.08.2012 / acceptance: 10.09.2012
Conflicts of interest: None
EXPECTING THE MOST UNEXPECTED – A HARLEQUIN BABY! A CASE REPORT AND LITERATURE ANALYSIS
Sundaramoorthy M. Srinivasan
Chettinad Hospital and Research Institute, Kelambakkam, Tamilnadu, India
Corresponding author: Prof. Sundaramoorthy Srinivasan e-mail: hamsrini@yahoo.co.in
How to cite an article: Srinivasan SM. Expecting the most unexpected – a harlequin baby! A case report and literature analysis. Our Dermatol Online. 2012; 3(4):321-325.
Abstract
Twenty eight years old primi gave birth to an amazing live baby by vaginal delivery – a Harlequin. The child was born with massive, diamond-shaped scales which limit the child’s movements. The term harlequin comes from the baby’s facial expression and diamond-shaped pattern of the scales like the 17th century entertainers, harlequins. As the skin cracks at places of movement the protective function of the skin is lost. Till now the prenatal diagnosis was based on biopsy of fetal skin carried out in an advanced stage of pregnancy. The most important indication of fetal cutaneous biopsy is the diagnosis of genodermatosis and hereditary diseases including Harlequin ichthyosis. It is predictable that for Harlequin Ichthyosis the prenatal test based on DNA will replace the fetal biopsy made before tenth week of gestation by chorionic villus sampling, or even before, with non-invasive analysis of the DNA of fetal cells in maternal circulation. Advances in neonatal intensive care along with scaling being made easier by the use of systemic retinoids has led to improved survival and the use of the name “Harlequin baby” to “Harlequin fetus”, “World’s Largest Baby” or “World’s Smallest Baby”. The mortality rate for harlequin ichthyosis is high. With neonatal intensive care and the advent of retinoid therapy, some babies have survived the newborn period. They are still at risk of dying from systemic infection. It’s the world’s most unconquired medical challenge.
Key words: harlequin; ichthyosis; eclabium; ectropion; ABCA12; lipid transport
Introduction
The term harlequin comes from the baby’s facial expression and diamond-shaped pattern of the scales like the 17th century entertainers, harlequins. As the skin cracks at places of movement the protective function of the skin is lost. Till now the prenatal diagnosis was based on biopsy of fetal skin carried out in an advanced stage of pregnancy. The most important indication of fetal cutaneous biopsy is the diagnosis of genodermatosis and hereditary diseases including Harlequin ichthyosis. It is predictable that for Harlequin Ichthyosis the prenatal test based on DNA will replace the fetal biopsy made before tenth week of gestation by chorionic villus sampling, or even before, with non-invasive analysis of the DNA of fetal cells in maternal circulation. Advances in neonatal intensive care along with scaling being made easier by the use of systemic retinoids has led to improved survival and the use of the name “Harlequin baby” to “Harlequin fetus”, “World’s Largest Baby”, “World’s Smallest Baby”, „Polymelia”, „Diphallia”, „Anencephaly”, „Ectopia Cordis”, „Harlequin ichthyosis”, „Craniopagus parasiticus”, „Cyclopia”.
Diprosopus are the world`s ten most amazing babies.
The mortality rate for harlequin ichthyosis is high. With neonatal intensive care and the advent of retinoid therapy, some babies have survived the newborn period. They are still at risk of dying from systemic infection. It’s the world’s most unconquired medical challenge.
Case Report
A 28-year-old non consanguineous married woman ,a primi, delivered a full term live male baby at term per vaginum, weighing 2450 g, with APGAR scores of 6, 7 and 8 at 1, 5, and 10 minutes respectively. I was called to examine the baby which was abnormal. When I went there, to my amazement, the baby was a Harlequin. The infant had characteristics typical of Harlequin Ichthyosis (HI) (Fig. 1, 2) a parchment-like appearance of the skin, ectropion, eclabium and low-set dysplastic ears. There was flexion deformity of all joints of the limbs. The hands and feet were swollen.
There was no prenatal identification of Harlequin Ichthyosis and there was no family history of the disease. Prenatally Ultrasound examination revealed no evident fetal abnormalities, except mild oligohydraminos.An optimal anomaly screening could not be performed due to oligohydramnios. Despite the oligohydramnios, the fetal movement pattern appeared grossly unrestricted.
The infant was transported to the neonatal intensive care unit and an umbilical vein catheter was inserted. Fluids were given intravenously as partial parenteral nutrition and protection from dehydration. After appropriate cultures were taken, prophylactic antibiotic treatment was started and intensive skin care involving moisturizing and oiling with baby olive oil. Ophthalmic lubricants were used to protect the conjunctivae. Despite this intensive care, respiratory insufficiency and anuria began on the fourth day of follow-up. Culture of skin swabs revealed Candida and Staphylococcus. Despite optimum treatment with catecholamine, antibiotics and ventilator support, and neonatologist`s care the infant died due to sepsis on the fifth day.
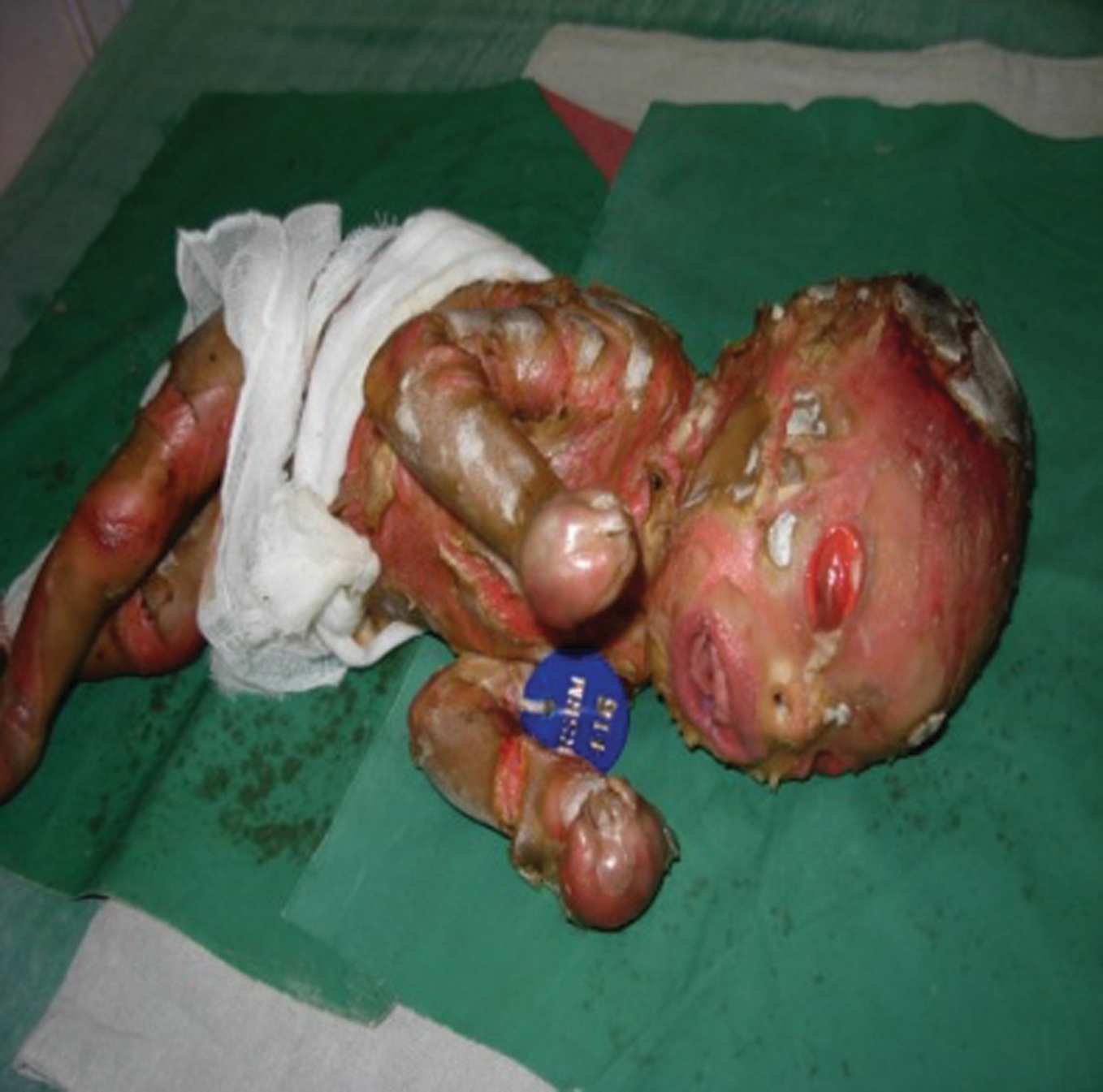 Figure 1. Harlequin Baby immediately after delivery – lateral view
|
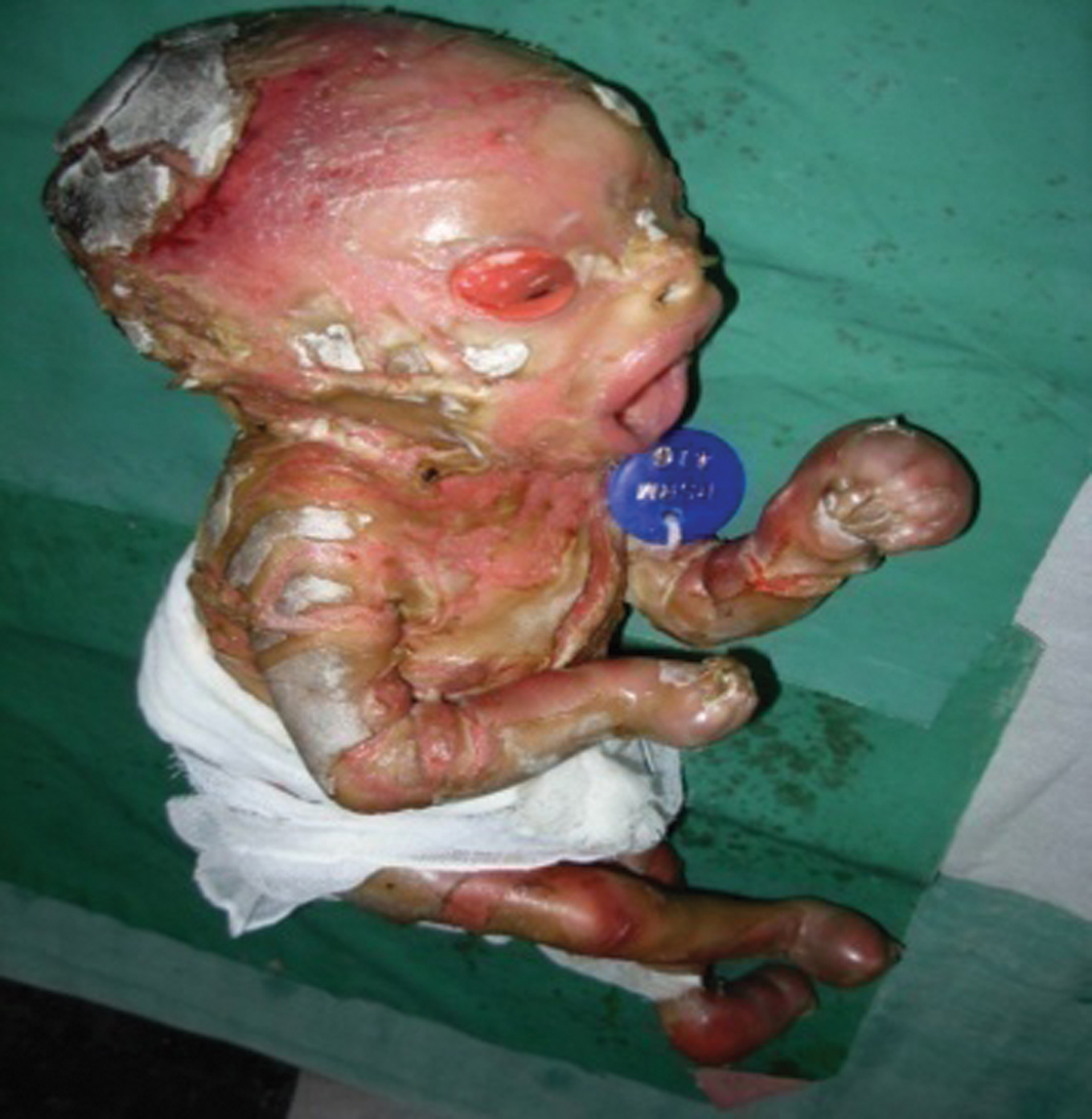 Figure 2. Harlequin Baby – anterolateral view
|
Discussion
In Harlequin infants, premature birth is typical, leaving infants at risk for additional complications from early delivery. These infants are also at high risk for difficulty breathing, infection, low body temperature, and dehydration. Constriction and swelling of the mouth may interfere with the suck response and infants may need tube feeding. Medical monitoring is difficult because of the abnormal skin; electrodes cannot be placed effectively and blood vessels cannot be seen under the skin. Placing lines in the artery and vein of the umbilical cord can aid in monitoring the infant and delivering fluids and nutrition. These infants may have problems maintaining normal levels of electrolytes, especially sodium in their blood. They are particularly prone to develop hypernatremia (high sodium levels in the blood). The baby’s corneas need to be lubricated and protected if the eyelids are forced open by the tightness of the skin. A high humidity environment in a heated incubator is necessary to help maintain body temperature and to prevent the skin from cracking. Antibiotic treatment may be necessary to prevent infection at this time. Administration of a retinoid such as oral etretinate1 (1 mg./kg. body weight) may accelerate shedding of the thick scales. Most Harlequin infants will need one-on-one nursing care for the first several weeks of life. In the past, Harlequin infants rarely survived the first few days of life. However, with recent advances in neonatal care and perhaps with the administration of etretinate,1 Harlequin infants can survive. Several surviving children with Harlequin ichthyosis are now young adults. The surviving children display dry, reddened skin, which may be covered by large thin scales, and sparse hair. Physical development may be delayed by the enormous calorie needs their skin function demands, but their mental and intellectual developments are expected to be normal. Harlequin ichthyosis demands a meticulous ongoing skin care regimen to keep the skin moisturized and pliable and to prevent cracking and fissuring that may lead to infection. Harlequin ichthyosis is a recessively inherited genetic disorder. In order to express (show) the disorder, individuals must inherit two recessive genes, one from each parent, but the parents (the „carriers”) show no signs. Recently, the cause of harlequin ichthyosis was traced to the ABCA12 gene. The ABCA12 gene is believed to encode a transporter protein involved in the transport of epidermal lipids across cell membranes. Identification of this gene has made DNA-based prenatal diagnosis of harlequin ichthyosis possible. Harlequin ichthyosis (HI) – the most severe form of keratinizing disorders, often lethal in the neonatal period is characterized by a profound thickening of the keratin skin layer, a dense “armor”-like scale that covers the body, and contraction abnormalities of the eyes, ears, and mouth. Akiyama et al. [21,22,23,24,25] report that mutations in ABCA12 caused defective lipid transport that significantly impacted normal development of the skin barrier. Lipid secretion was recovered after corrective ABCA12 gene transfer into patient keratinocytes. These results should allow for early prenatal diagnosis of HI and lend hope to the possibility of a specific treatment for this devastating disorder. Waring pointed to what is believed to be the first harlequin fetus described in the US in the diary of Reverend Oliver Hart in 1750 [1].
Pathogenesis
Lipid Lipid processing in the skin is essential for the protective function of the stratum corneum, the most external layer of the epidermis. Corneocytes, attached to each other by corneo-desmosomes and embedded in intercellular lipid lamellae, form a cornified layer that acts as a barrier between the internal and external environment for bodily defense. The lipid lamellae are derived from lamellar granules, the major lipid-rich organelles present in epidermal granular cells, which originate from the trans-Golgi network. At the granular layer–stratum corneum interface, the lamellar granules fuse with the cell membrane and discharge their content into the intercellular lamellae. Complex enzymatic reactions lead to modifications of the lipid composition of the intercellular space (cholesterol, ceramides, and free fatty acids) that provide a very effective water-permeability barrier. Corneocytes detach from each other in the superficial layers of the stratum corneum as a result of finely regulated proteolytic cleavage of corneo-desmosomes. In the skin of HI patients, the absence of ABCA12 prevents the transfer of lipids into lamellar granules, which themselves are abnormally shaped, reduced in number, or absent. As a result, exocytosis of lamellar granule content is reduced and intercellular lipid lamellae are absent. Abnormal lipid-containing vacuoles form in the cytoplasm of the corneocytes. The stratum corneum is remarkably thickened and does not desquamate. In a previous study, electron microscopy in HI patients revealed that lamellar granules are either absent or abnormal and that no intercellular lamellae are present. These data suggest that this defect in the lamellar granules results in thickening of the stratum corneum and the accumulation of armor-like scales in HI. However, the genetic basis for these events had not been elucidated. This discovery of the role of ABCA12 in HI reveals a major role of lipid transport in the formation of the skin barrier and its function. This involves two closely related lipid transporters, ABCA3 and ABCA12, which are essential for the production of alveolar surfactant and lipid lamellae in the stratum corneum, respectively. At birth, while ABCA3 prevents the lungs from collapsing, ABCA12 protects the skin from external aggressions and water loss. Loss of ABCA12 expression results in the most severe dysregulation of cornification in humans, covering the newborn infant in a lethal type of armor. Not only will these findings dramatically improve our ability to offer mutational screening and early DNA-based prenatal diagnosis of HI, but they will also allow for the development of new and specific therapeutic approaches. Genetic correction of ABCA12 deficiency by gene transfer in patients’ keratinocytes restored normal glucosylceramide cell distribution and lamellar granule formation. This result raises the possibility of HI treatment using systemic administration of functional peptides with ABCA12-like properties or ABCA12 gene delivery approaches undertaken either prior to or after birth [10,11,12].
Differential Diagnosis
Gaucher’s disease – beta glucocerebrosidase defect [13].
X linked recessive ichthyosis – Steroid sulfatase defect [9].
Niemann – Pick disease – Sphingomyelinase defect [14].
Sjogren-Larsson syndrome – Fatty aldehyde dehydrogenase defect [15].
Autosomal recessive congenital ichthyosis – Lipoxygenase-3 and 12R- lipoxygenase defect [16].
Dorfman – Chanarin syndrome – CGI-58 defect [17].
Tangier disease – Defect in cholesterol transport between liver and other tissues [2,3,4,5].
Stargardt disease – Abnormal accumulation of retinoid [6,7,8].
Neu-Laxova syndrome – Autosomal recessive disorder with severe IUGR, extreme microcephaly, ichthyosis, marked edema with skin restriction, cranio facial anomalies, limb deformities and CNS malformations.
Conclusion
HI is a rare and extremely severe form of congenital ichthyosis, with an incidence of about 1 in 300 000 births. Prenatal diagnosis is usually difficult because of non-specific signs in the ultra sonographic examination and rareness of the disorder. Delivery of a child with congenital ichthyosis identifies a family at risk, and for subsequent pregnancies prenatal diagnosis can be offered. This report is a typical example of all of these issues.
The ten most amazing babies in the world medical literature are:
1. World’s largest baby
2. World’s smallest baby
3. Polymelia
4. Diphallia
5. Anencephaly
6. Ectopia cordis
7. Harlequin ichthyosis
8. Craniophagus parasiticus
9. Cyclopia
10. Diprosopus.
According to the literature survey the most constant sonographic findings are: a large gaping mouth; dysplastic or swollen hands and feet; aplasia of the nose; and bulging eyes. The “snowflake sign” reflecting skin particles floating in the amniotic cavity, intra-amniotic debris or floating membranes might be another indirect sign (Tabl. I) [18,19,20]. Polyhydramnios has been proposed as a marker of congenital ichthyosis [18]. However, we detected oligohydramnios in our case, and oligohydramnios was also present in the cases reported in the literature. In the first half of the pregnancy, the contribution of water transport across the highly permeable skin of the fetus to the amniotic fluid volume is a major mechanism of amniotic fluid dynamics. At 22 to 25 weeks of gestation, keratinization of the skin occurs; it is generally accepted that significant amounts of water and solute are not transferred across this membrane after keratinization. We could not demonstrate the cause of oligohydramnios in our case; neither any abnormal Doppler result nor any structural malformation or preterm premature rupture of membranes was detected. Definite prenatal diagnosis of HI is based upon the examination of fetal skin biopsy samples obtained after 19-23 weeks’ gestation. Samples taken from the scalp will give a surer early diagnosis but are more difficult and more risky [20]. The use of three-dimensional ultrasound diagnosis does not appear to have been widely adopted, though useful. In HI mortality is very high that most affected neonates die within a few days due to sepsis, respiratory failure, infections, poor nutrition, and electrolyte imbalances. In 2003, Berg et al. could find only nine reported cases of prolonged survival of HI cases. Prolonged survival with better neonatal care is possible with developments of neonatal care and targeted oral retinoid therapy. Isotretinoin, etretinate, and acitretin were used for treatment of HI to achieve survival up to 8 years. Sing et al. successfully treated neonate with 1mg/kg/ day (started 10 days after birth) up to 30 months. Though survival rates appear to be increasing, the severity of the persisting dermatosis results in a lifetime of suffering for the saved individuals. This makes early prenatal diagnosis to allow appropriate counseling that is highly desirable for the parents. Prenatal diagnosis of HI remains difficult and is still usually only feasible in cases where there has already been an affected child. Better prenatal diagnostic procedures are needed.
|
Constant sonographic findings
|
|
Persistent large gaping mouth
|
|
Dysplastic or swollen hands and feet
|
|
Aplasia of the nose
|
|
Bulging eyes
|
|
Indirect signs
|
|
“ Snow flake sign”
|
|
Intra- amniotic debris
|
|
Floating membranes
|
|
Non specific ultrasonographic signs
|
|
Polyhydramnios
|
|
Oligohydramnios
|
Table I. Most constant ultrasonographic signs of Harlequin ichthyosis
UNANSWERED QUESTIONS
1. Do we have the technical knowhow and skills to diagnose prenatally/intrauterine of HI?
2. Do we have medical technology to give treatment intrauterine for HI? Is it feasible? Is it advisable? What sort of research going on regarding this condition?
3. Is it possible to make sustainable correction of dermatological defects soon after birth or neonatally?
4. As this case is an interdisciplinary approach, is the role of the dermatologist defined?
5. Is it medico legally accepted or any legislations executed for MTP if diagnosed prenatally?
6. What is the possibility of another child having same disease? Is there any medical records highlighting the same?
7. What sort of medical counseling should be given?
Finally there are many questions than answers for this condition….!!!
REFERENCES
1. Waring JJ: Early mention of a Harlequin fetus in America. Am J Dis Child. 1932;43:442. 2. Bodzioch M, Orsó E, Klucken J, Langmann T, Böttcher A, Diederich W, et al: The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347- 51. 3. Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, et al: Mutations in ABC1 in tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336-45. 4. Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, et al: Tangier disease is caused by mutations in the gene encoding ATPbinding cassette transporter 1. Nat Genet. 1999;22:352-5. 5. Remaley AT, Rust S, Rosier M, Knapper C, Naudin L, Broccardo C, et al: Human ATP-binding cassette transporter 1 (ABC1): genomic organization and identification of the genetic defect on the original Tangier disease kindred. Proc Natl Acad Sci U S A. 1999;96:12685-90. 6. Martínez-Mir A, Paloma E, Allikmets R, Ayuso C, del Rio T, Dean M, et al: Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18:11-2. 7. Rozet JM, Gerber S, Ghazi I, Perrault I, Ducroq D, Souied E, et al: Mutations of the retinal specific ATP binding transporter gene (ABCR) in a single family segregating both autosomal recessive retinitis pigmentosa RP19 and Stargardt disease: evidence of clinical heterogeneity at this locus. J Med Genet. 1999;36:447-51. 8. Cremers FP, van de Pol DJ, van Driel M, den Hollander AI, van Haren FJ, Knoers NV, et al: Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene. ABCR Hum Mol Genet. 1998;7:335- 62. 9. Webster D, France JT, Shapiro LJ, Weiss R: X-linked ichthyosis due to steroid-sulphatase deficiency. Lancet. 1978;1:70-2. 10. Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, et al: Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115:1777-84. 11. Kelsell DP, Norgett EE, Unsworth H, Teh MT, Cullup T, Mein CA, et al: Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet. 2005;76:794-803. 12. Lefévre C, Audebert S, Jobard F, Bouadjar B, Lakhdar H, Boughdene-Stambouli O, et al: Mutations in the transporter ABCA12 is associated with lamellar ichthyosis type 2. Hum Mol Genet. 2003;12:2369-78. 13. Sidransky E, Tsuji S, Martin BM, Stubblefield B, Ginns EI: DNA mutation analysis of Gaucher patients. Am J Med Genet. 1992;42:331-6. 14. Schmuth M, Man MQ, Weber F, Gao W, Feingold KR, Fritsch P, et al: Permeability barrier disorder in Niemann-Pick disease: sphingomyelin-ceramide processing required for normal barrier homeostasis. J Invest Dermatol. 2000;115:459-66. 15. De Laurenzi V, Rogers GR, Hamrock DJ, Marekov LN, Steinert PM, Compton JG, et al: Sjogren-Larsson syndrome is caused by mutations in the fatty aldehyde dehydrogenase gene. Nat Genet. 1996;12:52-7. 16. Jobard F, Lefèvre C, Karaduman A, Blanchet-Bardon C, Emre S, Weissenbach J, et al: Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum Mol Genet. 2002;11:107-13. 17. Lefèvre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, et al: Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet. 2001;69:1002-12. 18. Berg C, Geipel A, Kohl M, Krokowski M, Baschat AA, Germer U, et al: Prenatal sonographic features of Harlequin ichthyosis. Arch Gynecol Obstet. 2003;268:48-51. 19. Holden S, Ahuja S, Ogilvy-Stuart A, Firth HV, Lees C: Prenatal diagnosis of Harlequin ichthyosis presenting as distal arthrogryposis using three-dimensional ultrasound. Prenat Diagn. 2007;27:566-7. 20. Montague I, Fox R, Mann R: Intra-amniotic debris identified at ultrasound scanning: a feature of congenital ichthyosis. Ultrasound Obstet Gynecol. 1997;9:350-1. 21. Akiyama M: Pathomechanisms of harlequin ichthyosis and ABCA transporters in human diseases. Arch Dermatol. 2006;142:914–8. 22. Akiyama M, Dale BA, Smith LT, Shimizu H, Holbrook KA: Regional difference in expression of characteristic abnormality of harlequin ichthyosis in affected fetuses. Prenat Diagn. 1998;18:425–36. 23. Akiyama M, Kim D-K, Main DM, Otto CE, Holbrook KA: Characteristic morphologic abnormality of harlequin ichthyosis detected in amniotic fluid cells. J Invest Dermatol. 1994;102:210-3. 24. Akiyama M, Sakai K, Wolff G, Hausser I, McMillan JR, Sawamura D, et al: A novel ABCA12 mutation 3270delT causes harlequin ichthyosis. Br J Dermatol. 2006;155:1064-6. 25. Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, et al: Mutations in ABCA12 in harlequin ichthyosis and functional rescue by corrective gene transfer. J Clin Invest. 2005;115:1777-84.
Comments are closed.