DOI: 10.7241/ourd.20124.74 article in PDF
Our Dermatol Online. 2012; 3(4): 336-341
Date of submission: 24.05.2012 / acceptance: 02.07.2012
Conflicts of interest: None
LESIONES TUMORALES MÚLTIPLES EN UN PACIENTE ADOLESCENTE
MULTIPLE TUMORAL LESIONS IN AN ADOLESCENT PATIENT
Beatriz Di Martino Ortiz1, Oilda Knopfelmacher1, Américo Sacco2
1Cátedra de Dermatología. Hospital de Clínicas. Facultad de Ciencias Médicas. Universidad Nacional de Asunción. Paraguay
2Práctica privada. Asunción. Paraguay
Corresponding author: Dr. Beatriz Di Martino Ortiz e-mail: beatrizdimartino@gmail.com
How to cite an article: Di Martino Ortiz B, Knopfelmacher O, Sacco A. Lesiones tumorales múltiples en un paciente adolescente. Our Dermatol Online. 2012; 3(4):335-340.
Resumen
Introduction: El xantogranuloma juvenil (XGJ) es una histiocitosis de células no Langerhans, benigna, autocurativa, que afecta con mayor frecuencia a lactantes y niños, que se caracteriza por pápulas amarillentas asintomáticas y/o nódulos situados en la piel y otros órganos y que consisten en un infiltrado de macrófagos con un variable grado de lipidización en ausencia de un trastorno metabólico. Presentamos el caso de un varón, adolescente, de 14 años de edad, con múltiples lesiones tumorales gigantes en brazos y pies, limitadas a la piel y que no requirieron tratamiento alguno.
Abstract
Introduction: Juvenile xanthogranuloma (JXG), a non-Langerhans cell histiocytosis, is benign and self-healing disorder, that affects most often infants and children, characterized by asymptomatic yellowish papules and/or nodules located in the skin and other organs consisting of an infiltrate of macrophages with a variable degree of lipidization in the absence of a metabolic disorder. We report the case of a male teenager of 14 years old, with multiple giant tumoral lesions in the arms and feet, limited to the skin and who did not require any treatment.
Palabras clave: xantogranuloma juvenil; histiocitosis de células no Langerhans; nevoxantoendotelioma
Key words: juvenile xanthogranuloma; non-Langerhans cells histiocytosis; nevoxanthoendoteliomais
Introducción
El XGJ se encuentra dentro de las histiocitosis de células no Langerhans o tipo II [1,2]. Recientemente, con el advenimiento de técnicas de inmunohistoquímica y estudios ultraestructurales, la Sociedad del Histiocito propuso reclasificar a las histiocitosis según el tipo celular predominante en 3 grupos:
1) Desórdenes derivados de células dendríticas (el cual incluiría a la histiocitosis de células de Langerhans y al XGJ entre otros),
2) Desórdenes derivados de macrófagos y
3) Enfermedades histiocíticas malignas.
El XGJ es una entidad tumoral, benigna y autolimitada que comienza típicamente en la infancia o juventud temprana con lesiones solitarias o múltiples.
Caso Clínico
Adolescente de 14 años de edad de sexo masculino, proviene de una zona rural del Paraguay. Consulta por lesiones sobre-elevadas en piel de brazo derecho y pies, de 7 años de evolución, que aumentan progresivamente de tamaño, no dolorosas, ni pruriginosas.
Antecedentes patológicos personales y familiares: sin datos de interés.
Examen Físico: Lesiones nodulares/tumorales de tamaños comprendidos entre 1.5 a 4.5 cm. de ejes mayores, de bordes regulares, y límites netos, cubiertas por piel normal, ligeramente amarillenta-anaranjada que afectan el brazo derecho (Fig. 1) y ambos pies (Fig. 2), donde se observan incluso algunas lesiones agrupadas. A la palpación son móviles, no adheridas a planos profundos y no son dolorosas. Algunas presentan telangiectasias en superficie (Fig. 3).
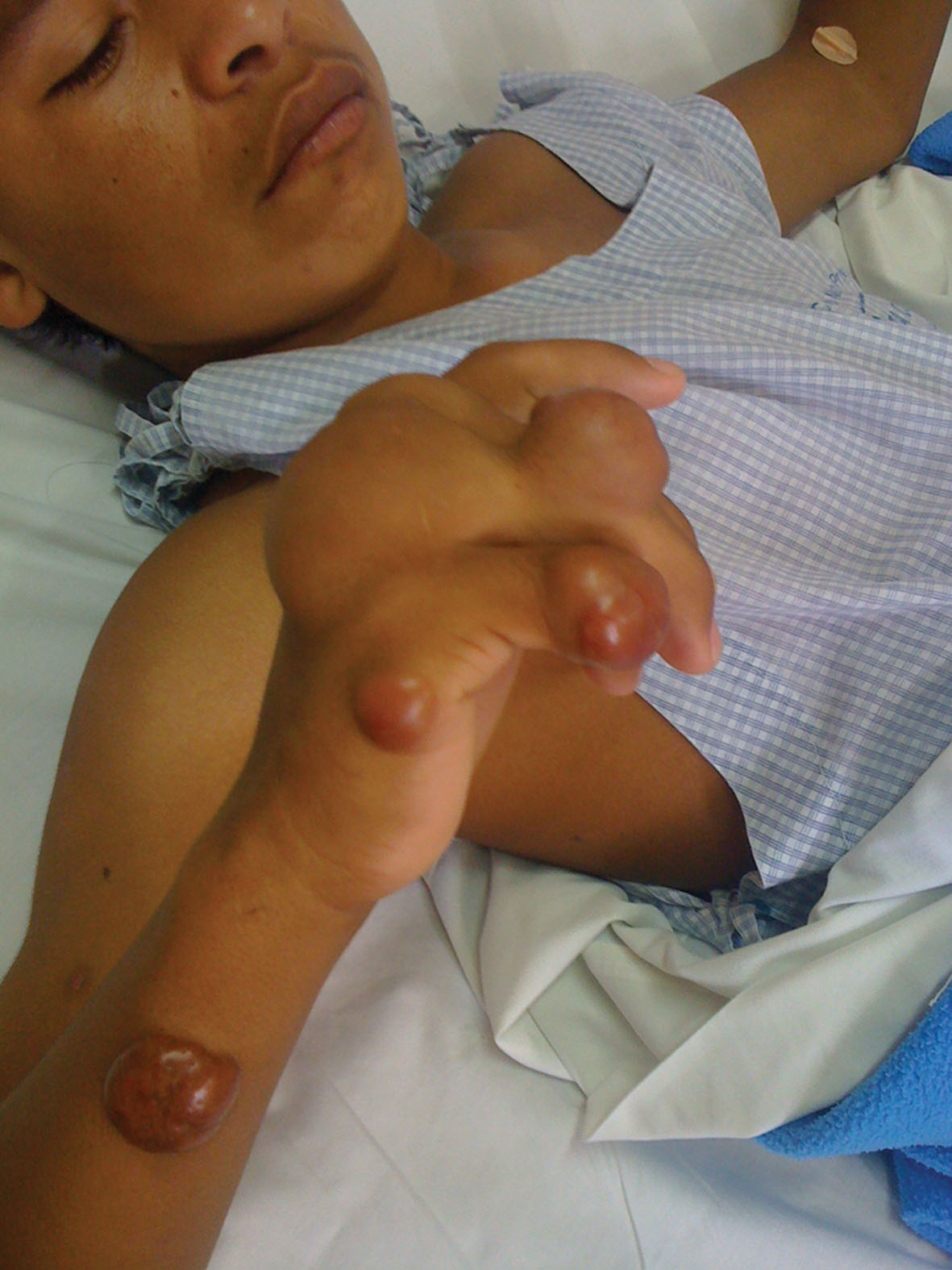 Figure 1. Clínica. Lesiones nodulares de tamaños comprendidos entre 1.5 a 4.5 cm. de ejes mayores, de bordes regulares, y límites netos, cubiertas por piel normal , ligeramente amarillenta-anaranjada que afectan el brazo derecho.
Figure 1. Clinic. Nodular lesions ranging in size from 1.5 to 4.5 cm. major axis, regular edges, and net limits, covered by normal skin, pale yellow-orange affecting his right arm.
|
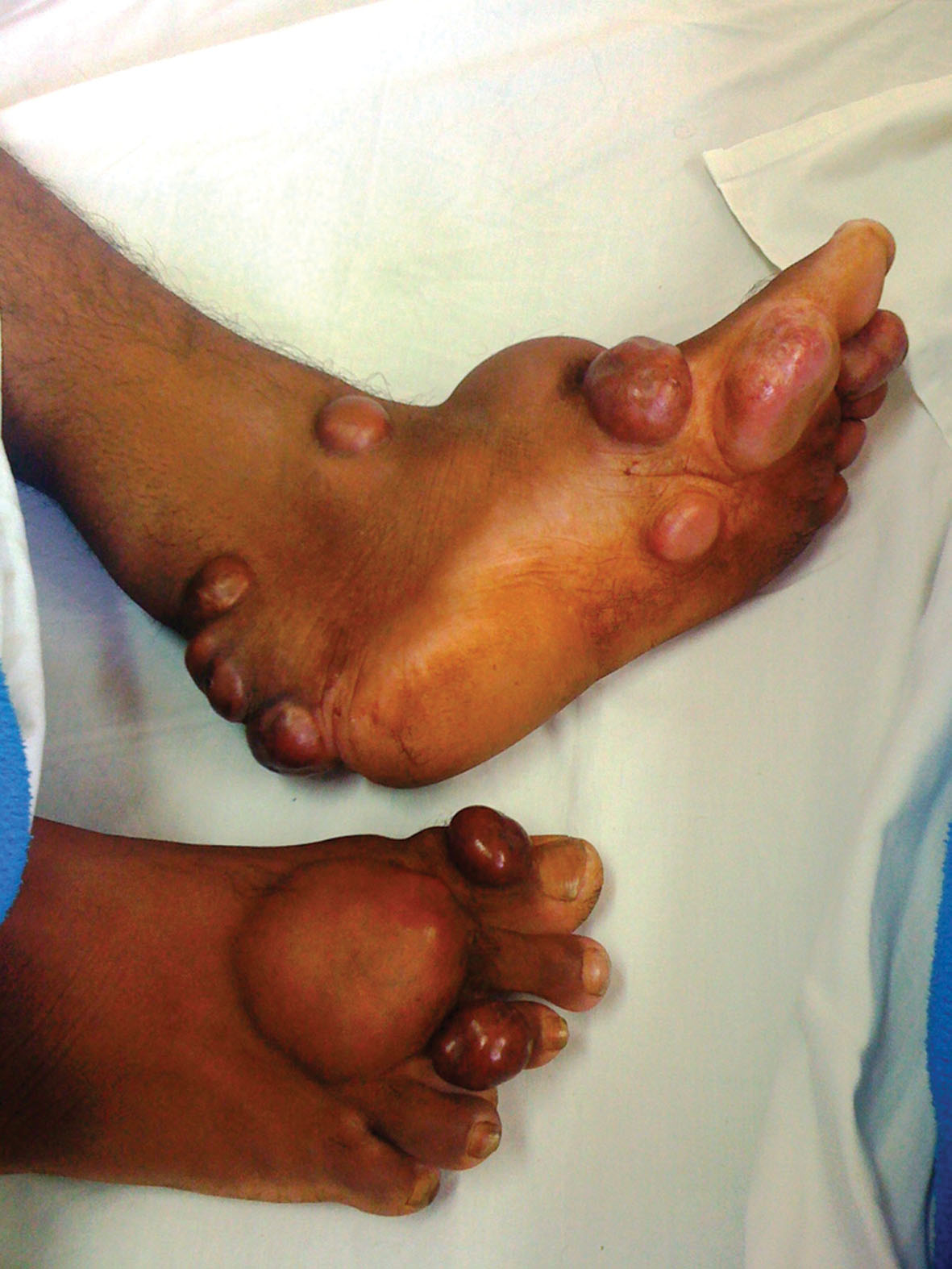 Figure 2. Clínica. Lesiones en ambos pies, donde se observan algunas agrupadas. A la palpación son móviles, no adheridas a planos profundos y no son dolorosas.
Figure 2. Clinic. Nodules in both feet, where there are agminated. On palpation are mobile, not attached to deeper layers and are not painful.
|
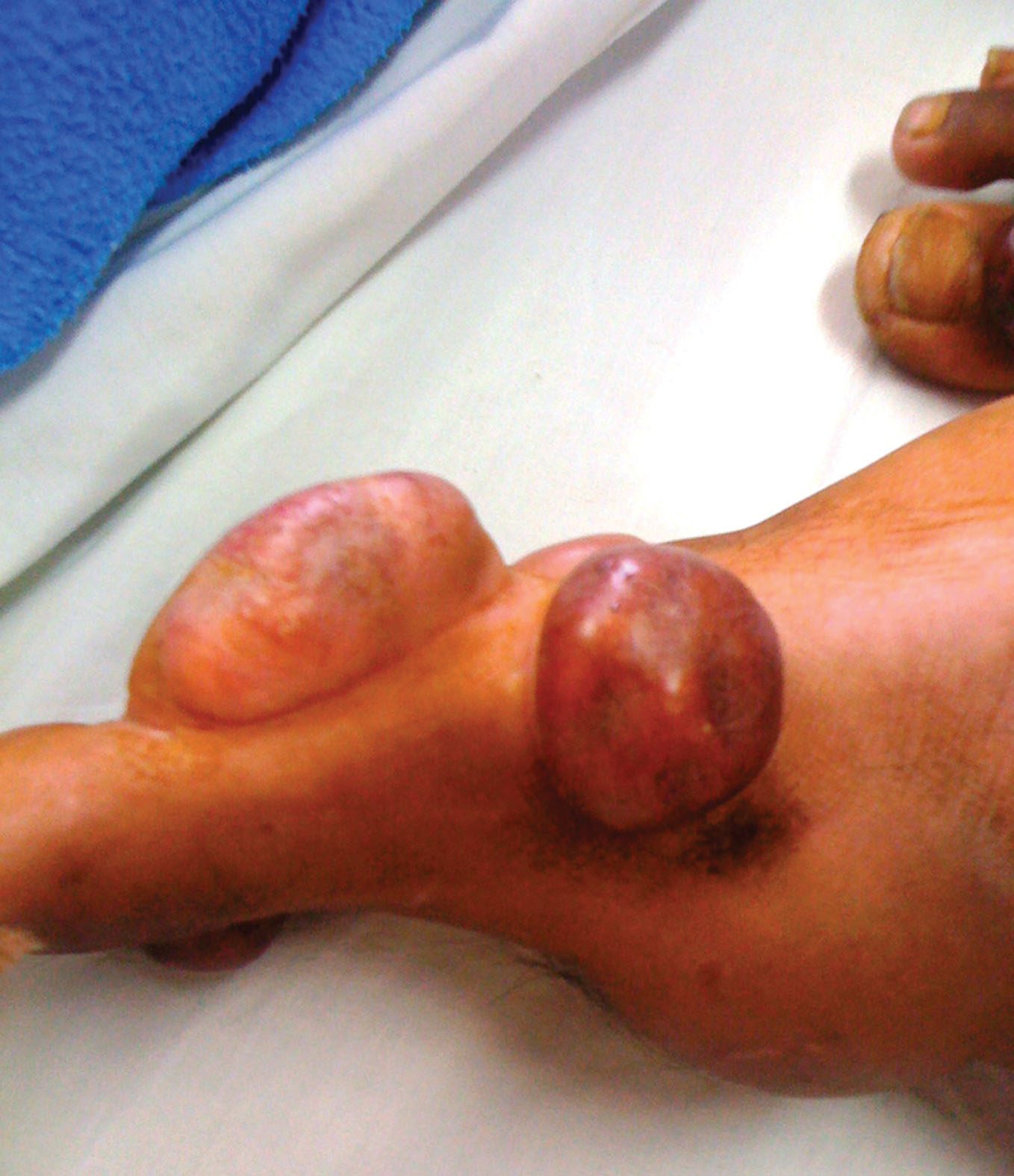 Figure 3. Clínica. Algunas presentan telangiectasias en superficie.
Figure 3. Clinic. Some have telangiectasias on the surface.
|
Auxiliares del diagnóstico:
· Hemograma, orina simple, perfil lipídio y perfil hepático: normales.
· Radiografías simples de miembros superiores o inferiores: afectación exclusiva de partes blandas. Ninguna afectación ósea.
· Examen oftalmológico: normal.
· Ecografía abdominal: normal.
Se efectúa extirpación quirúrgica de una lesión situada en el brazo y se remite a anatomía patológica, fijada en formol neutro tamponado al 10% y se procesa de manera rutinaria.
Anatomía patológica:
· Macroscopía: se recibe un fragmento cutáneo en cuña de 3 x 2.5 x 1.5 cm. de ejes mayores, sobre la que asienta una lesión nodular, de bordes netos y límites precisos, cubierta por piel no erosionada ni ulcerada. Al corte, se observa una tumoración amarilla homogénea, sin áreas de necrosis o hemorragia y es de consistencia sólida (Fig. 4).
· Microscopía: denso infiltrado dérmico, que respeta la epidermis de cual se halla separado nítidamente, compuesto por células espumosas, células gigantes de tipo cuerpo extraño y gigantes de Touton (Fig. 5), distribuidas principalmente en la porción más superficial de la lesión y en los bordes. No se encontraron linfocitos, eosinófilos, neutrófilos ni células plasmáticas dispersas a lo largo de la lesión. No se encontró fibrosis, y los lípidos no están presentes extracelularmente. La epidermis por encima de la lesión mostraba cierta atrofia con pérdida de redes de crestas e hiperpigmentación de la capa basal.
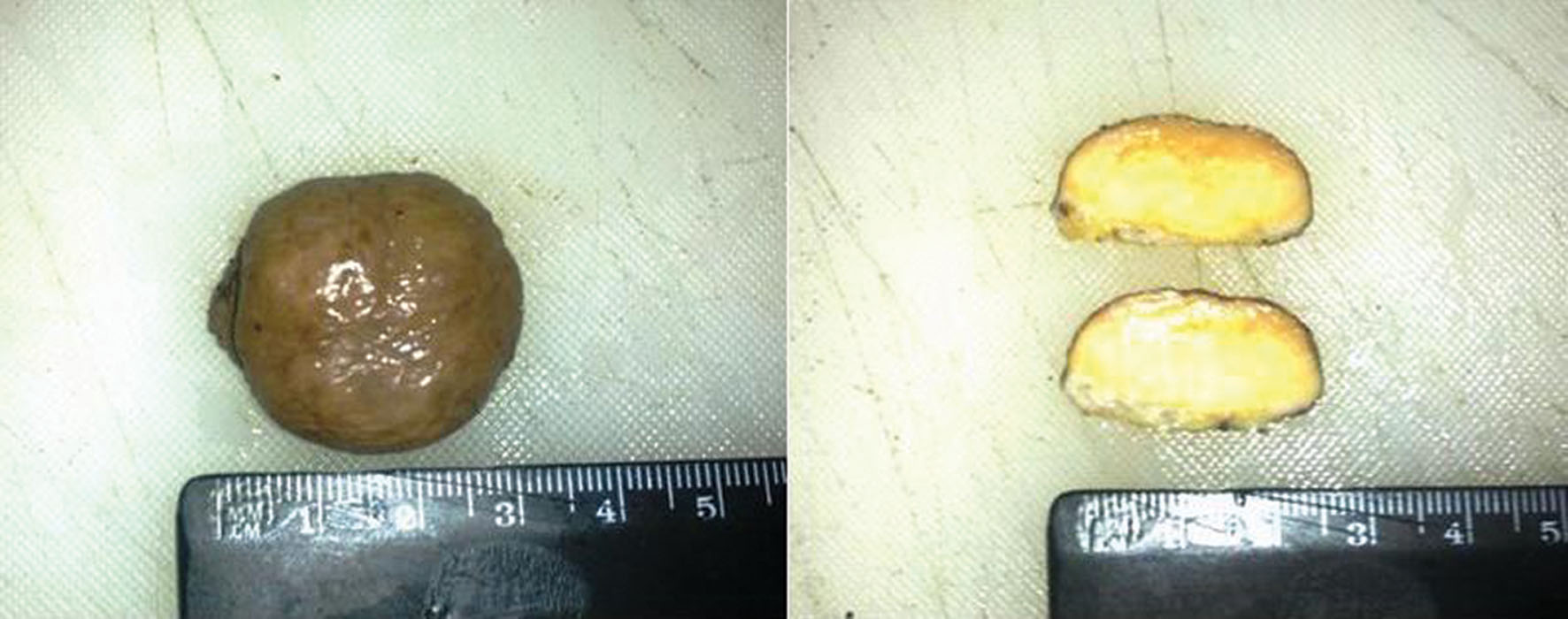 Figure 4. Histopatología. Fragmento cutáneo en cuña de 3 x 2.5 x 1.5 cm. de ejes mayores, sobre la que asienta una lesión nodular, de bordes netos y límites precisos, cubierta por piel no erosionada ni ulcerada (izquierda). Al corte, se observa una tumoración amarilla homogénea, sin áreas de necrosis o hemorragia y es de consistencia sólida (derecha).
Figure 4. Histopathology. Skin fragment of 3 x 2.5 x 1.5 cm. major axis, on which sits a nodular lesion of net edges and precise limits, covered by not eroded or ulcerated skin (left). Cut surface, yellow homogenous tumor without areas of necrosis or hemorrhage and solid consistency (right).
|
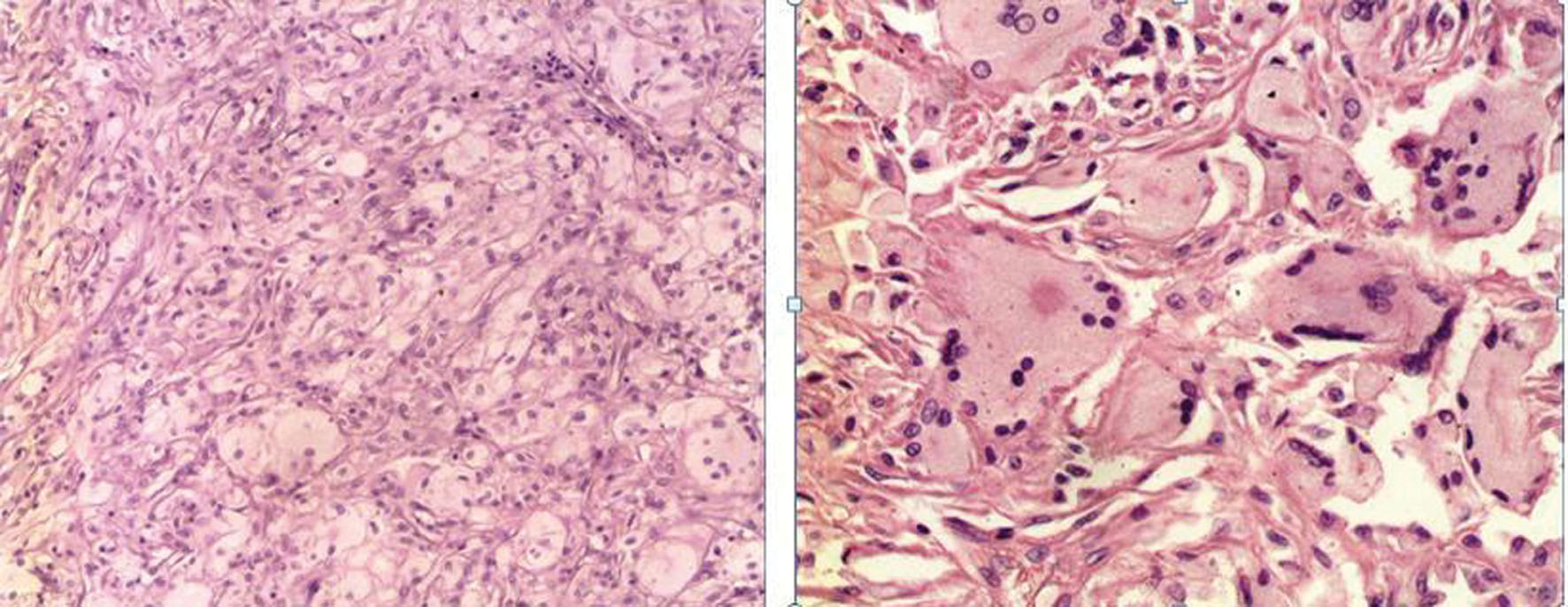 Figure 5. Histopatología. Denso infiltrado dérmico, compuesto por células espumosas (izquierda), células gigantes de tipo cuerpo extraño y gigantes de Touton (derecha).
Figure 5. Histopathology. Dense dermal infiltrate composed of foam cells (left), giant cells of foreign body and Touton type (right).
|
Diagnóstico Final:
Histiocitosis de células no langerhans (Tipo II): forma nodular gigante de xantogranulomas juveniles en fase proliferativa.
Conducta terapeútica asumida tras el diagnóstico anatomopatológico: conservadora con observación clínica.
Comentarios
El xantogranuloma juvenil (XGJ) fue descrito por Adamson [1] en 1905, quien lo llamó xantoma congénito múltiple. En 1954, Helwing y Hackney demostraron el origen fibrohistiocitico de la lesión y la denominaron xantogranuloma juvenil, término con el que se la conoce desde entonces. Las Histiocitosis tipo II o histiocitosis de células no Langerhans incluyen un grupo muy variado de enfermedades por proliferación de histiocitos cuyo fenotipo es diferente de la célula de Langerhans. Clínicamente se dividen en 3 grupos, las que afectan predominantemente la piel, otras que afectando la piel presentan una afectación sistémica predominante, y el tercer grupo de enfermedades que son principalmente extracutáneas. El prototipo del primer grupo es el xantogranuloma juvenil, y del tercer grupo la linfohistiocitosis hemofagocítica. Con el advenimiento de técnicas de inmunohistoquímica y estudios ultraestructurales, la Sociedad Internacional del Histiocito [3] propuso reclasificar a las mismas según el tipo celular predominante en 3 grupos: 1) Desórdenes derivados de células dendríticas (el cual incluiría a la histiocitosis de células de Langerhans y al XGJ entre otros), 2) Desórdenes derivados de macrófagos, y 3) Enfermedades histiocíticas malignas. Las clasificaciones se muestran en las tablas 1 y 2.
El XGJ tiene múltiples denominaciones: nevoxantoendotelioma, xantoma múltiple, xantoma juvenil, xantoma múltiple de la infancia, xantoma tuberoso congénito de la infancia, xantoma neviforme y granuloma juvenil de células gigantes. Su etiología es desconocida. Se sugiere que se produce aumento de la biosíntesis de colesterol intracelular y de la unión de colesterol a lipoproteínas de baja densidad (LDL) en ausencia de trastorno metabólico. Los resultados de algunos autores sugieren que los histiocitos en las lesiones XGJ tienen diferenciación macrofágica, que probablemente representa un proceso reactivo a un estímulo desconocido [11]. La incidencia es desconocida. Es más frecuente en caucásicos. La relación en lactantes es V:M=1,5:1 y en adultos V=M, para los XGJ solitarios. Para los XGJ múltiples la relación V:M=5:1.
|
Histiocitosis tipo I: Histiocitosis de células de Langerhans
|
|
Histiocitosis tipo II: Histiocitosis de fagocitos mononucleares distintos a las células de Langerhans
– Histiocitosis cefálica benigna, Xantogranuloma juvenil, Xantoma disseminatum,
Xantoma papular, Xantoma eruptivo generalizado, Reticulohistiocitoma.
– Linfohistiocitosis hemofagocítica (familiar y reactiva).
– Histiocitosis sinusal con linfadenopatía masiva (enfermedad de Rosai-Dorfman).
|
|
Histiocitosis tipo III: Histiocitosis malignas.
– Leucemia aguda monocítica (FAB M5).
– Histiocitosis maligna.
– Linfoma histiocitario verdadero.
|
Tabla I. Clasificación de las histiocitosis (Sociedad del Histiocito)
Table I. Classification of Histiocytosis (Histiocyte Society)
| ENFERMEDADES DE COMPORTAMIENTO BIOLÓGICO VARIABLE |
| Enfermedades de células dendríticas |
|
– Histiocitosis de células de Langerhans
– Enfermedades secundarias de células dendríticas
– Xantogranuloma juvenil y enfermedades relacionadas
– Histiocitoma solitario de fenotipo dendrítico
|
|
ENFERMEDADES MALIGNAS
|
|
Enfermedades de los monocitos
|
|
– Leucemias (según clasificación FAB revisada)
– Leucemia aguda monocítica (FAB M5A y B)
– Leucemia aguda mielomonocítica (FAB M4)
– Leucemia crónica mielomonocítica
– Tumor monocítico extramedular (Sarcoma granulocítico de tipo monocítico)
|
|
Enfermedades de células dendríticas
|
|
– Sarcoma histiocítico de células dendríticas (localizado o diseminado)
– Según el fenotipo: Sarcoma de célula dendrítica folicular, de célula dendrítica interdigitante,
etc.
– Sarcoma histiocítico de células macrofágicas (localizado o diseminado)
|
Tabla II. Clasificación actual de las enfermedades histiocíticas (adaptado del grupo de trabajo de la OMS y de la Sociedad del Histiocito)
Table II. Current classification of histiocytic diseases (adapted from the working group of WHO and of the Histiocyte Society)
Las lesiones aparecen en el primer año de vida en 60 a 75% de los casos. Son congénitos entre el 5 y 20%. Los adultos se afectan en 10 a 15%. Las lesiones son múltiples en 20 a 30% de los casos. No se han observado casos familiares. En cuanto a las manifestaciones clínicas no suelen comprometer el estado general, las lesiones cutáneas y viscerales tienden a la auto resolución, en el trascurso de 3-6 años, siguiendo un curso benigno y de buen pronóstico.
Las variantes clínicas principales son:
1. Micronodular o Papular (60%): pápula o nódulo amarillo, anaranjado o rosado, de 2 a 5 mm de diámetro frecuentemente localizada en la parte superior del cuerpo, y
2. Macronodular o Nodular (35%): tumores de 1 a 2 cm de diámetro, con telangiectasias en la superficie y con mayor frecuencia de afección mucosa y sistémica. Esta forma se ve más frecuentemente en adultos.
Las variantes poco usuales son: mixtas, gigante (término que se reserva para los nódulos que superan los 2 cm. de dinámetro [10], frecuentes en escápula y en nariz (Cyran), subcutáneas (lesión solitaria, congénita, profunda, > 2cm.) y otras (agrupadas, liquenoide, generalizada, máculo papular, reticulada, apareada). En cuanto a la localización las lesiones cutáneas están irregularmente dispersas a lo largo de la piel sin una tendencia al agrupamiento, y se encuentran principalmente en la parte superior del cuerpo. Las membranas mucosas rara vez pueden estar afectadas. La manifestación extracutánea más común del XGJ (que se presenta principalmente en la forma papular, en niños <2 años, en formas múltiples y con formas subcutáneas) es la afectación ocular. Las lesiones oculares pueden ocurrir en aproximadamente en el 1-10% de los niños y son casi siempre unilaterales pudiendo producir efecto masa, glaucoma, hifema e incluso ceguera. Las lesiones oculares pueden preceder o seguir a las manifestaciones cutáneas. En estos casos, podría estar indicada la revisión oftalmológica cada 6 meses hasta los 2 años de edad. La variante nodular de XGJ de vez en cuando puede estar relacionada con manifestaciones sistémicas de los pulmones, los huesos, los riñones, pericardio, colon, ovarios, testículos y sistema nervioso central. Se han descripto asociaciones de XGJ con enfermedades mieloproliferativas y también con la neurofibromatosis tipo I (NF1), entre otras [4]. La enfermedad mieloproliferativa más frecuentemente hallada es una variante de leucemia mielomonocítica o leucemia mieloide cronica tipo juvenil (LMC), con mala evolución y desenlace fatal en la mayoría de los pacientes [5]. La forma papular, que es la más frecuente y que se caracteriza por numerosas (hasta 100) lesiones firmes, son las que se pueden asociar en quizás un 20% de los pacientes con manchas café-au-lait de la neurofibromatosis y pueden estar relacionadas con leucemia mieloide crónica juvenil Histopatológicamente se describen tres estadios evolutivos: Inicial (inicio de la proliferación histiocitaria, con prácticamente nula cantidad de células gigantes), Proliferativo (al observarse células espumosas o células de Touton), y Cicatrizal (cuando se evidencia fibrosis) [9]. La inmunomarcación de estos tumores demuestra positividad para CD45, CD68 y FXIII [11]. Casos S100+ no excluyen el diagnóstico de XGJ. El origen dendrocito dérmico de estos tumores se plantea en base a la positividad para FXIII (lo cual no explica la proliferación xantogranulomatosa en sitios extracutáneos). Casos CD4+ se han utilizado como evidencia de que los monocitos plasmáticos pueden ser el tipo celular constituyente principal de XGJ, en lugar del dendrocito dérmico [12]. Los hallazgos inmunohistoquímicos de las células histiocíticas se muestran en la Tabla III.
|
Progenitor hematopoyético CD34+
|
Precursores
cutáneos
mesenquimales
(fibroblásticos)
|
|
IHQ
|
Macrófago |
Célula
indeterminada
|
Célula de
Langerhans
|
Dendrocito |
|
CD45
|
+
|
+
|
+
|
+
|
|
CD68
|
+
|
–
|
+
|
– |
|
FXIII
|
–
|
–
|
–
|
+ |
| S100 | – | + | + | -/+ |
| CD1a | – | + | + | – |
|
Gránulo de Birbeck
|
–
|
–
|
+
|
–
|
Table III. Inmunomarcación de células histiocitarias
Table III. Immunostaining of histiocytic cells
En cuanto al tratamiento suele ser conservador ya que tienden a la auto resolución en 3 a 6 años dejando hipopigmentación, anetodermia o leve atrofia cutánea. El estado de salud del niño no está afectado y su desarrollo físico y psicomotor es normal, salvo si hay complicaciones por su localización visceral o por asociación con otras enfermedades. Se describen algunos casos evolución fatal, pero éstos han tenido manifestaciones del SNC o hígado. El cribado de una patología visceral o hematológica asociada debe estar guiado por la clínica y no debe realizarse indiscriminadamente en todos los pacientes con una forma aislada de XGJ. Se recomiendo sobre todo dentro de los 2 primeros años, hemogramas periódicos, ya que es la edad donde se observa el mayor pico de incidencia de la LMC juvenil. El tratamiento es conservador, adoptando una actitud expectante, dado que el curso es benigno y autolimitado, sobre todo en las formas múltiples o en las gigantes (como en nuestro caso), evitando así las secuelas estéticas de la intervención. La biopsia es necesaria ante duda diagnóstica, siendo en formas papulares y micronodulares (<2 cm), además, terapéutica. Las acciones terapéuticas dependerán de los síntomas y complicaciones que ocasione. Cuando existe compromiso sistémico se debe realizar un seguimiento estrecho y sólo se tratan si modifican signos vitales. Los XGJ multisistémicos pueden requerir tratamiento con corticoides y/o quimioterapia. Se puede requerir cirugía. En cuanto al compromiso ocular exclusivo, pueden realizarse, según la gravedad del caso, tratamientos quirúrgicos, radioterapia, corticoides sistémicos o locales [6,7].
Conclusión
El XGJ es una enfermedad predominantemente de la edad pediátrica, benigna y autolimitada, con compromiso exclusivo de la piel en la mayoría de los casos, y que no requiere tratamiento alguno. Sin embargo deben realizarse todos los exámenes complementarios a los pacientes, con el fin de identificar compromiso extra cutáneo, asociaciones y evitar las posibles complicaciones. El interés de comunicar este caso reside en que se trata de una variante clínica inusual (forma nodular gigante) de una condición común [8]. Esta patología por lo general es de aparición congénita o en los primeros meses de vida y en nuestro caso se ha iniciado en un niño mayor. Las lesiones cutáneas se localizan principalmente en cara, cuello, tronco superior y, con menos, frecuencia en raíz de las extremidades, encontrándose en nuestro caso afectación de la porción inferior del cuerpo, inclusive.
REFERENCES
1. Zelger BW, Sidoroff A, Orchard G, Cerio R: Non-Langerhans cell histiocytoses. A new unifying concept. Am J Dermatopathol. 1996;5:490-04.
2. Sidwell RU, Francis N, Slater DN, Mayou SC: Is disseminated juvenile xanthogranulomatosis benign cephalic histiocytosis? Pediatr Dermatol. 2005;22:40-3.
3. Satter EK, High WA: Langerhans cell histiocytosis: A review of the current recommendations of the Histiocyte Society. Pediatr Dermatol. 2008;25:291-6.
4. Magnin PH, Marini MA, Oxilia MR: Asociación de xantogranuloma juvenil y neurofibromatosis de von Recklinghausen. Seguimiento de un caso durante 21 años. Rev Argent Dermatol. 1985;66:209-13.
5. Pérez Wilson J, de Moragas JM: El xantogranuloma juvenil y su asociación con la neurofibromatosis. Revisión de la literatura. Dermatologia (Chile). 1990;6:82-5.
6. Shaw M, Costantini SE, Pérez MB, Abulafia J yet al: Xantogranuloma juvenil. Arch Argent Dermatol. 1984;34:91-9.
7. Buján MM, Sosa G, Cervini AB, Laterza A, Pierini AM: Xantogranuloma juvenil: experiencia en un hospital pediátrico. Dermatol Argent. 2010;16:262-7.
8. Kaur MR, Brundler MA, Stevenson O, Moss C: Disseminated clustered juvenile xanthogranuloma: an unusual morphological variant of a common condition. Clin Exp Dermatol. 2008;33:575-7.
9. Ngendahandao P, de Saint Aubain N: Mitotically Active Xanthogranuloma: A Case Report With Review of the Literature. Am J Dermatopathol. 2012;34:27-30.
10. Avechu Diaz A, Navarro González D, Virto Ruiz MT: Xantogranuloma juvenil gigante. An Pediatr (Barc). 2012;76:300-1.
11. Sangüeza OP, Salmon JK, White CRJr, Beckstead JH: Juvenile xanthogranuloma: a clinical, histopathologic and immunohistochemical study. J Clin Pathol. 1995;22:327-35.
12. Kraus MD, Haley JC, Ruiz R, Essary L, Moran CA, Fletcher CD: „Juvenile” xanthogranuloma: an immunophenotypic study with a reappraisal of histogenesis. Am J Dermatopathol. 2001;23:104-11.
Comments are closed.